Caso clínico - Hepatología
←
→
Transcripción del contenido de la página
Si su navegador no muestra la página correctamente, lea el contenido de la página a continuación
Caso clínico
Deficiencia de lipasa ácida lisosomal, una
enfermedad subdiagnosticada. Reporte de caso
Lysosomal acid lipase deficiency, an
underdiagnosed disease. Case report
Suad Siuffi-Campo1 , Ricardo Londoño-García1 , Yeinis Paola Espinosa-Herrera2 ,
Juan Camilo Pérez-Cadavid 3
, Octavio G. Muñoz-Maya 4
,
Resumen
La deficiencia de lipasa ácida lisosomal (LAL-D) es una enfermedad rara de herencia autosómica
recesiva, causada por mutaciones en el gen LIPA, localizado en el cromosoma 10 (10q23.31),
la cual causa el acúmulo sistémico y progresivo de ésteres de colesterol y triglicéridos. Se han
reportado más de 40 mutaciones en dicho gen, por lo cual las manifestaciones clínicas de la enfer-
medad son diversas, predominando la hepatopatía y la enfermedad cardiovascular de aparición
temprana. Se han descrito pocos casos a nivel mundial de esta enfermedad. En este reporte se
expone el caso de un paciente con LAL-D, quien inicialmente fue tratado como glucogenosis. Más
tarde, se confirmaron las alteraciones en los lípidos séricos, la deficiencia de la enzima, así como
la mutación correspondiente a dicha deficiencia enzimática.
Palabras clave: lipasa, metabolismo de los lípidos, enfermedad de acumulación de colesterol éster,
dislipidemia, hepatomegalia, cirrosis, hígado graso.
Abstract
Lysosomal acid lipase deficiency (LAL-D) is a rare disease of autosomal recessive inheritance, caused by
mutations in the LIPA gene, located on chromosome 10 (10q23.31), which causes systemic and pro-
gressive accumulation of cholesterol esters and triglycerides. More than 40 mutations in this gene have
been reported, with diverse clinical manifestations, with hepatic disease and early-onset cardiovascular
disease being the predominant ones. Few cases of this condition have been described worldwide. This
report presents the case of a patient with LAL-D, who was initially treated for glycogenosis. Later, the
alterations in serum lipids, the enzyme deficiency, as well as the corresponding mutation to the enzyme
deficiency were confirmed.
1
Médicos Generales, Hospital Pablo Tobón Uribe. Medellín, Colombia.
2
Médica, Residente de Medicina Interna, Universidad de Caldas. Manizales, Colombia.
3
Médico, Especialista en Patología, Hospital Pablo Tobón Uribe. Ayudas Diagnósticas SURA. Profesor, Facultad de Medicina,
Universidad Pontificia Bolivariana. Medellín, Colombia.
4
Médico, Especialista en Medicina Interna, Especialista en Hepatología Clínica y Trasplante Hepático. Hospital Pablo Tobón
Uribe. Universidad de Antioquia. Medellín, Colombia. E-mail: octavio.g.munoz@gmail.com.
Conflicto de interés: los autores declaran que no tienen conflicto de interés.
Hepatología 2022;3:97-105. https://doi.org/10.52784/27112330.151.
Recibido el 25 de agosto de 2021; aceptado el 24 de octubre de 2021. Editora Médica Colombiana S.A., 2022©.
Volumen 3 | Número 1 | Ene-Jun 2022 97
|Siuffi-Campo S, Londoño-García R, Espinosa-Herrera YP, Pérez-Cadavid JC, Muñoz-Maya OG
Keywords: acid lipase, lipid metabolism, cholesterol ester storage disease, dyslipidemia, hepatome-
galy, cirrhosis, steatohepatitis.
Introducción sustancias antes descritas y se acumulan
en los lisosomas, ocasionando una regula-
La deficiencia de lipasa ácida lisosomal ción al alza en la producción de la enzima
(LAL-D) es una enfermedad de herencia HMG-CoA reductasa, obteniendo como
autosómica recesiva que hace parte de resultado final un aumento en los niveles
las enfermedades de depósito lisosomal. de colesterol libre y triglicéridos [6,7].
Su incidencia y prevalencia son raras
y se caracteriza por la acumulación de La acumulación progresiva de los ésteres
ésteres de colesterol y triglicéridos en el de colesterol y de los triglicéridos en los
hígado, bazo y otros órganos [1]. La in- lisosomas se manifiesta clínicamente me-
cidencia se ha estimado según algunos diante hepatoesplenomegalia, esteatosis
estudios genéticos en 1/40.000 en la hepática, fibrosis y/o cirrosis hepática,
población alemana [2]. Las formas más que pueden desencadenar una insuficien-
severas presentan una incidencia cercana cia hepática. Aun cuando las manifestacio-
a 1/500.000, como lo reflejan estudios nes clínicas pueden ser muy variables, por
en Australia [3]. Esta enfermedad afecta lo general se presenta aumento de la alani-
por igual a uno u otro sexo y se ha repor- na aminotransferasa (ALT), lipoproteínas de
tado con mayor frecuencia en niños, con baja densidad (LDL), lipoproteínas de alta
un pico de incidencia entre los 3 y 15 densidad (HDL) y triglicéridos [1,8,9]. La
años [4]. Es una enfermedad heterogénea dislipidemia es entonces un común denomi-
de espectro clínico variable dentro de los nador en estos pacientes, y por lo tanto, las
individuos que la presentan [1]. Se cono- personas que padecen esta enfermedad
ce que la LAL-D se produce por mutaciones pueden sufrir de aterosclerosis avanzada
en el gen LIPA ubicado en el cromosoma [1,8,9]. Las enfermedades cardiovascula-
10 (10q23.31), que está conformado por res constituyen la primera causa de muerte
10 exones y tiene aproximadamente 45 en estos pacientes [10]. Generalmente,
kb de longitud [3]. A la fecha, se han iden- los pacientes con inicio de la enfermedad
tificado más de 40 mutaciones de pérdida en el periodo neonatal presentan un curso
de la función [3,4]. Esta enfermedad apa- clínico más agudo comparado con los ni-
rece habitualmente en personas homocigo- ños y adultos. La presentación infantil se
tas o heterocigotas compuestas para las describió en 1956, y recibió históricamen-
mutaciones en el gen LIPA [4]. Las mutacio- te el nombre de enfermedad de Wolman
nes de tipo nonsense, frameshift o puntua- [11]. Como síntomas iniciales aparecen
les son las más severas y son detectadas el vómito, diarrea, falla del crecimiento y
en los niños, mientras que las mutaciones hepatoesplenomegalia, produciendo un
menos severas ocurren en los adultos [4]. rápido deterioro de la función hepática y
La lipasa ácida lisosomal es clave en el fallecimiento en promedio a los 3,7 meses
metabolismo de los lípidos, pues hidroliza [12]. En aproximadamente un 50% de to-
los ésteres de colesterol y los triglicéridos dos los recién nacidos y niños, se pueden
en los lisosomas [5]. Por lo tanto, cuando observar calcificaciones en las glándulas
la actividad de esta enzima está ausente o adrenales en las imágenes radiológicas
reducida, no pueden ser degradadas las [1,3,13,14]. Posteriormente, se desarro-
98 Deficiencia de lipasa ácida lisosomal, una enfermedad subdiagnosticada. Reporte de caso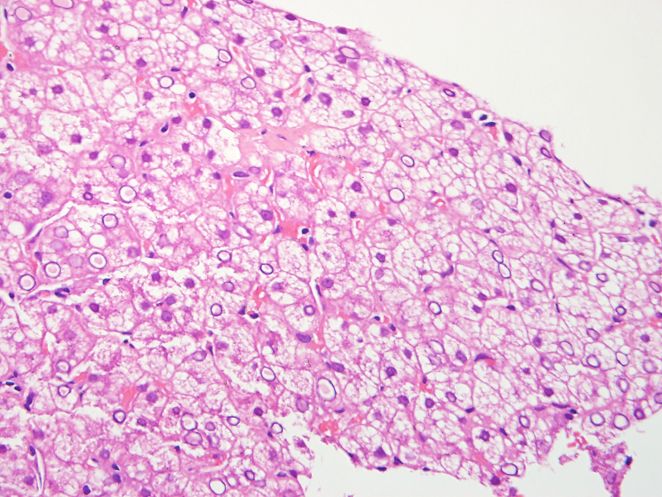
llan manifestaciones a causa de fallas mul- crecimiento. Consultó inicialmente por
tiorgánicas, particularmente falla hepática cuadro clínico consistente en hepatome-
con ictericia y caquexia [3]. La historia na- galia, elevación de las transaminasas y
tural de la enfermedad en niños y adultos dislipidemia. Se realizó en el 2003 una
está menos definida [15], pero se conoce primera biopsia hepática con reporte his-
que, aproximadamente, un tercio de los tológico compatible con glucogenosis.
pacientes experimentan síntomas gastroin- Estuvo en seguimiento y controles desde
testinales severos, incluyendo diarrea, vó- el 2007, con mejoría de la hepatome-
mito, distensión abdominal, malabsorción galia y la función hepática. Sin embar-
y esteatorrea [1,15,16]. go, no tuvo diagnóstico molecular has-
ta agosto del 2016, cuando se realizó
La LAL-D es un reto diagnóstico, pues hay ExomeNext-Trio™ en ambos padres y
un desconocimiento de la existencia de paciente. Sin embargo, en ese momen-
esta enfermedad, y los signos y síntomas to no se pudo identificar ninguna varian-
se parecen a otras condiciones clínicas te previamente descrita en la literatura
más comunes como hipercolesterolemia consultada, ni en las bases de datos de
familiar, esteatohepatitis no alcohólica, variantes asociadas a patología que se
enfermedad del hígado graso no alcohó- relacionen como una causal al fenotipo
lico, enfermedad de Niemann Pick, enfer- del paciente en estudio. Posteriormente,
medad de Gaucher o cirrosis criptogénica en agosto del 2018, se realizó un estu-
[1,8,17-20]. dio de microarreglos (CMA, del inglés,
Chromosomal Microarray) en la Clínica
A continuación, expondremos un caso Mayo, donde se demostraron deleciones
clínico de un paciente con hepatopatía y alteraciones en el cromosoma 10, no
y dislipidemia, que por muchos años se relacionadas con glucogenosis. Finalmen-
trató como una glucogenosis. Después te, en enero del 2019, con un estudio
de mucho tiempo se pudo diagnosticar multigen para enfermedades de almace-
como LAL-D, siendo entonces el ejemplo namiento del glucógeno, se descartaron
perfecto de lo que esta enfermedad repre- cambios asociados a glucogenosis, y de
senta como reto diagnóstico, al mimetizar forma incidental se encontró homocigosis
hepatopatías más comunes, y por ende, del gen LIPA para déficit de lipasa ácida
conduce a que esta enfermedad esté sub- lisosomal. La actividad reportada de la
diagnosticada y se tenga un desconoci- enzima fue de 31,2 mcmol/L/h (valor de
miento global sobre el impacto real de la referencia: >32,5 mcmol/L/h). Para el
enfermedad. Es de importancia realizar seguimiento de este paciente, se decidió
este reporte de caso, pues podría aportar estudiar el grado de hepatopatía median-
información, no solo sobre las manifesta- te realización de biopsia hepática (figu-
ciones clínicas, sino también sobre pautas ras 1 a 4), se suspendió el tratamiento
para el abordaje diagnóstico. con fécula de maíz, y se remitió a las
diferentes especialidades para estudiar el
Caso clínico grado de afectación cardiovascular. Ac-
tualmente, el paciente está recibiendo tra-
Se trata de un paciente de género mascu- tamiento interdisciplinario con Nutrición,
lino, conocido a los 4 años y 10 meses Hepatología y Cardiología. El pilar de
de edad en el Hospital Pablo Tobón Uri- su tratamiento son las estatinas para el
be. Como único antecedente patológico control del riesgo cardiovascular, y se le
presentó retraso en el desarrollo pondoes- ha insistido en hábitos de vida saludable
tatural, con suplencia de hormona del y en la realización de ejercicio.
Volumen 3 | Número 1 | Ene-Jun 2022 99
|Siuffi-Campo S, Londoño-García R, Espinosa-Herrera YP, Pérez-Cadavid JC, Muñoz-Maya OG
Figura 4. Tinción de catepsina D por
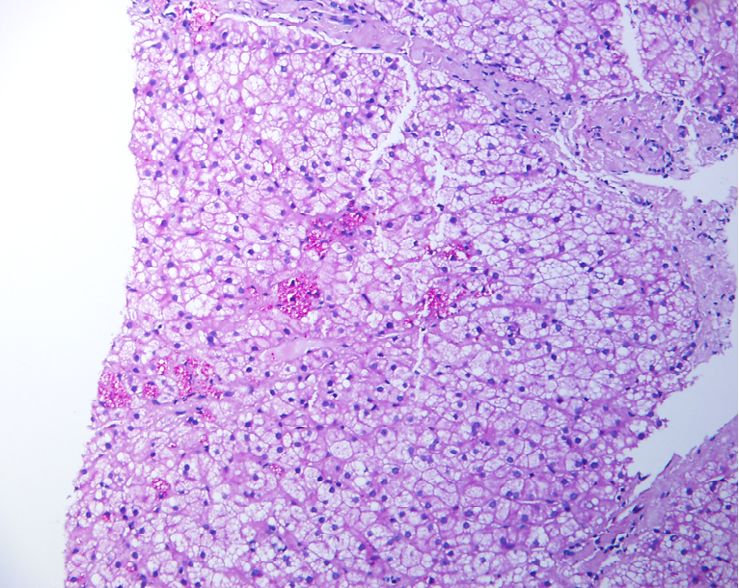
Figura 1. Parénquima hepático con células inmunohistoquímica (200x). Marcación positiva
hepatocitarias microvacuoladas y núcleos de las microvacuolas lipídicas en el citoplasma
glucogenizados, H&E (400x). de los hepatocitos, con mayor intensidad en los
macrófagos.
Discusión
La LAL-D tiene un amplio rango de ma-
nifestaciones fenotípicas, que pueden ir
desde la presentación infantil temprana,
aparición durante la infancia o en etapas
tardías de la vida. En el caso de nuestro
paciente, inició con síntomas antes de los
5 años de vida, y las características que
Figura 2. Tinción de tricrómico (400x). Se describimos en este reporte coinciden con
resaltan focos de fibrosis pericelular. la variabilidad que tiene la presentación
de la enfermedad. Desde muy temprano
tenía peso y talla bajos, además de he-
patomegalia, hipercolesterolemia, hiper-
trigliceridemia, y AST y ALT elevadas. La
hepatomegalia con o sin esplenomegalia
es frecuente, y está presente en el 99% de
los niños y en el 74% de los adultos con
LAL-D [2]; este fue uno de los hallazgos
que se encontraron en las valoraciones ini-
ciales. El hígado es el órgano más afecta-
do en la fisiopatología de la enfermedad,
los niveles elevados de ALT y AST pueden
ser el indicio temprano del daño hepático
que puede manifestarse con o sin icteri-
cia, esteatosis, fibrosis o cirrosis, y llevar a
Figura 3. Tinción de PAS (200x). Hay ausencia complicaciones asociadas como várices
de captación de glucógeno en el citoplasma esofágicas, progresar a insuficiencia o fa-
de los hepatocitos. Resaltan los macrófagos
lla hepática aguda, cirrosis o carcinoma
cargados con microvacuolas lipídicas.
hepatocelular [21].
100 Deficiencia de lipasa ácida lisosomal, una enfermedad subdiagnosticada. Reporte de casoAhora bien, se trata de una enfermedad diagnóstico, y son un rasgo característico
que resulta ser un reto diagnóstico, no de LAL-D en niños y adultos. La presencia
solamente por el poco conocimiento que de marcadores lisosomales luminales y de
se tiene de esta, sino también porque mi- membrana alrededor de las vacuolas lipídi-
metiza muchas otras hepatopatías, y por- cas es indicativa de LAL-D, al igual que los
que el abordaje diagnóstico es difícil y cristales de éster de colesterol [25-28]. En
requiere técnicas moleculares complejas y cuanto a las imágenes diagnósticas, una
de tecnología de punta. Como parte de resonancia magnética hepática con espec-
los métodos diagnósticos, se encuentran troscopia ha sido recientemente propuesta
los estudios de laboratorio. El diagnóstico como método diagnóstico no invasivo y de
de LAL-D puede realizarse a través de la seguimiento [29].
medición de la actividad de la enzima
lipasa ácida o por la determinación de Desde los primeros reportes por Abramov,
mutaciones en el gen LIPA [22]. De hecho, Schorr y Wolman, al observar en un lac-
esta enfermedad puede ser confirmada tante acúmulos de ésteres de colesterol y
bioquímicamente al medir la actividad de triglicéridos en hígado, bazo y glándulas
la enzima en un cultivo de fibroblastos, leu- suprarrenales, se estableció como uno de
cocitos periféricos o tejido hepático. Sin los pilares del tratamiento el cambio en el
embargo, los sustratos, como por ejem- estilo de vida, y el manejo con estatinas por
plo el 4-nitrophenyl palmitate, utilizados su acción hipolipemiante al disminuir los ni-
en estos ensayos, pueden no ser especí- veles de LDL; no obstante, estas no generan
ficos, haciendo que puedan generarse cambios en el desarrollo de la fibrosis he-
falsos negativos. Por eso, surge un nuevo pática [11,29,30]. Entre otros tratamientos
método para determinar la actividad de novedosos se describe la vitamina E [31].
LAL en sangre seca, utilizando el substra- Por su lado, el trasplante de hígado y el
to 4-methylumbelliferyl palmitate [22,23]. hematopoyético también han sido consi-
Mediante una secuenciación completa de derados como alternativas cuando no se
las regiones codificantes para el gen LIPA, logra una estabilidad en el progreso de la
se puede realizar una caracterización del enfermedad [32,33]. Se han venido intro-
estado genético de los individuos a quie- duciendo nuevos tratamientos con base a
nes se les sospecha LAL-D [3]. Aunque la la sustitución enzimática, para restablecer
mutación más común es la E8SJM, esta se los niveles fisiológicos normales de la enzi-
presenta en un 50% a 70% de los alelos ma y así prevenir la acumulación de éste-
mutados en los niños y adultos con LAL-D res de colesterol y triglicéridos, al retrasar
[24], lo que dificulta el diagnóstico cuan- la progresión de la enfermedad [8,25]. La
do no se presenta en el porcentaje restante sebelipasa alfa es una terapia enzimática
de personas que padecen la enfermedad. recombinante humana estudiada como te-
rapia para LAL-D [1]. Un estudio en fase
La biopsia hepática se considera un mé- 1-2 en adultos con LAL-D demostró que la
todo confiable para orientar sobre la etio- sebelipasa alfa se asoció con un rápido de-
logía. Es común el reporte de esteatosis cremento de las aminotransferasas, acom-
microvesicular, pero no es exclusivo de pañado por un mejoramiento en el perfil
LAL-D, por lo que se necesitan otros signos lipídico [34,35]. El estudio ARISE (del in-
histológicos para confirmar el diagnóstico. glés, Acid Lipase Replacement Investigating
La presencia de histiocitos, aumento en el Safety and Efficacy) fue un estudio de fase
tamaño de las células de Kupffer con in- 3, multicéntrico, doble ciego, comparado
cremento de vacuolas, gotas de lípidos y con placebo, que incluyó a 66 pacientes
cristales de colesterol, pueden apoyar el con estadio avanzado de la enfermedad
Volumen 3 | Número 1 | Ene-Jun 2022 101|Siuffi-Campo S, Londoño-García R, Espinosa-Herrera YP, Pérez-Cadavid JC, Muñoz-Maya OG
(ALT 1,5 veces por encima del límite supe- y ecografía abdominal. Es opcional la
rior de la normalidad, LDL ≥190 mg/dL, realización de intervenciones adicionales
y el 30% de los pacientes con biopsia he- tales como biopsia hepática, fibroscan,
pática disponible presentaban cirrosis). Tras resonancia magnética, ecocardiografía
20 semanas de tratamiento con sebelipasa y ecografía de carótidas. Actualmente
alfa, hubo mejoría tanto en el nivel de tran- hay poco conocimiento sobre la utilidad
saminasas, como en el de LDL, con pocos de ciertos biomarcadores como los de
efectos adversos, los cuales fueron leves activación macrofágica (quitotriosidasa,
[18]. Wilson y colaboradores también de- quimiocinas o metabolitos oxidados del
mostraron mejoría en el perfil lipídico, con colesterol) o factores que pueden modi-
un patrón menos aterogénico tras 52 sema- ficar la expresión vascular del gen LIPA
nas de tratamiento con sebelipasa, al com- (homocisteína, interleuquina-6 o el factor
pararla con la administración de placebo, V Leyden) [38]. Sin tratamiento, puede
tanto en niños como en adultos con LAL-D haber progresión de la enfermedad he-
[36]. Se requerirán estudios adicionales pática con aparición de fibrosis y progre-
para determinar si los cambios en el curso sión a cirrosis. La dislipidemia crónica no
natural de la enfermedad son completos o tratada puede conducir a aterosclerosis
resultarán en fenotipos emergentes de la en- acelerada y alto riesgo de complicacio-
fermedad [37]. nes cardiovasculares y cerebrovasculares
[37]. Se ha estimado que los pacientes
El objetivo del tratamiento de sustitución en- con LAL-D tienen un riesgo cardiovascular
zimática es el de restaurar la actividad de 54% mayor, a diez años, que la pobla-
la LAL, de manera cercana a la fisiológica, ción general [38].
y así evitar la progresión de la enfermedad,
independientemente del fenotipo de la en- Conclusiones
fermedad [38]. Se puede realizar la repo-
sición con enzima recombinante de lipasa LAL-D es una patología con una baja
ácida lisosomal humana (rhLAL) o sebelipa- prevalencia, posiblemente secundaria al
sa alfa recombinante [39]. Este fármaco es gran subdiagnóstico o diagnóstico erra-
una proteína glucosilada que se une a los do, lo que puede condicionar que pase
receptores manosa-6-fosfato de membrana desapercibida o se confunda con otras
plasmática, se internaliza mediante endo- enfermedades de depósito o que compro-
citosis y se trasporta a los lisosomas [38]. meten el hígado. Existen varios fenotipos
Una vez adentro, la enzima recombinante dependiendo de la edad de presentación
reemplaza a la LAL, hidrolizando ésteres de y del grado de actividad enzimática;
colesterol y triglicéridos dentro del lisosoma, cuando la actividad es mínima, la LAL-D
para revertir sus efectos [39]. Fue aproba- se manifiesta de manera temprana, con
da en Europa en 2015 para el tratamiento una mayor severidad y se asocia a mayor
de sustitución enzimática a largo plazo en mortalidad. En la mayoría de los casos
pacientes de todas las edades con LAL-D no se hace un diagnóstico oportuno o
[38]. El costo del medicamento sigue sien- no se sospecha esta entidad debido a la
do un desafío importante, particularmente falta de conocimiento, y también al acce-
para países con recursos de atención médi- so limitado de las pruebas diagnósticas
ca limitados [37]. confirmatorias, lo cual hace pensar que
la prevalencia real de la entidad es ma-
En cuanto al seguimiento y pronóstico de yor. En Colombia y Latinoamérica no se
la LAL-D, se recomienda la realización se- cuenta con estudios de prevalencia sobre
riada de pruebas hepáticas, perfil lipídico esta enfermedad.
102 Deficiencia de lipasa ácida lisosomal, una enfermedad subdiagnosticada. Reporte de casoLa LAL-D debe sospecharse ante la presen- 6. Horton JD, Goldstein JL, Brown MS. SREBPs:
cia de alteraciones hepáticas o lipídicas activators of the complete program of choles-
persistentes, sin causa clara, ante lo cual terol and fatty acid synthesis in the liver. J Clin
estaría indicado la determinación de la ac- Invest 2002;109:1125-1131. https://doi.
tividad de LAL y un posterior diagnóstico org/10.1172/JCI15593.
de confirmación con el análisis molecular 7. Cummings MH, Watts GF. Increased hepatic se-
del gen LIPA. Clásicamente, el tratamiento cretion of very-low-density lipoprotein apolipopro-
se ha basado en el control de la dislipi- tein B-100 in cholesteryl ester storage disease.
demia y el uso de fármacos hipolipemian- Clin Chem 1995;41:111-114.
tes, los cuales no mejoran el compromiso 8. Reiner Ž, Guardamagna O, Nair D, Soran H,
hepático. La sebelipasa alfa es una lipasa Hovingh K, Bertolini S, et al. Lysosomal acid
ácida lisosomal (LAL) humana recombinan- lipase deficiency--an under-recognized cause
te, aprobada para el tratamiento de susti- of dyslipidaemia and liver dysfunction. Athe-
tución enzimática, mejorando los paráme- rosclerosis 2014;235:21-30. https://doi.
tros lipídicos y hepáticos en pacientes con org/10.1016/j.atherosclerosis.2014.04.003.
esta enfermedad. El trasplante hepático es 9. Burton BK, Deegan PB, Enns GM, Guardamagna
la única alternativa posible en casos con O, Horslen S, Hovingh GK, et al. Clinical features
evolución a insuficiencia hepática terminal. of lysosomal acid lipase deficiency. J Pediatr Gas-
troenterol Nutr 2015;61:619-625. https://doi.
Referencias org/10.1097/mpg.0000000000000935.
10. Bernstein DL, Lobritto S, Iuga A, Remotti H,
1. Bernstein DL, Hülkova H, Bialer MG, Des- Schiano T, Fiel MI, et al. Lysosomal acid lipase de-
nick RJ. Cholesteryl ester storage disease: ficiency allograft recurrence and liver failure- clini-
review of the findings in 135 reported pa- cal outcomes of 18 liver transplantation patients.
tients with an underdiagnosed disease. J He- Mol Genet Metab 2018;124:11-19. https://
patol 2013;58:1230-1243. https://doi. doi.org/10.1016/j.ymgme.2018.03.010.
org/10.1016/j.jhep.2013.02.014. 11. Abramov A, Schorr S, Wolman M. Gene-
2. Pisciotta L, Fresa R, Bellocchio A, Pino E, ralized xanthomatosis with calcified adre-
Guido V, Cantafora A, et al. Cholesteryl nals. AMA J Dis Child 1956;91:282-286.
ester storage disease (CESD) due to no- https://doi.org/10.1001/archpe-
vel mutations in the LIPA gene. Mol Genet di.1956.02060020284010.
Metab 2009;97:143-148. https://doi. 12. Jones SA, Valayannopoulos V, Schneider E,
org/10.1016/j.ymgme.2009.02.007. Eckert S, Banikazemi M, Bialer M, et al. Ra-
3. Grabowski G, Charmas L, Du H. Lysosomal acid pid progression and mortality of lysosomal
lipase deficiencies: The Wolman disease/cho- acid lipase deficiency presenting in infants.
lesteryl ester storage disease spectrum. Metab Genet Med 2016;18:452-458. https://doi.
Mol Bases Inherit Dis 2012;142:1-31. org/10.1038/gim.2015.108.
4. Saito S, Ohno K, Suzuki T, Sakuraba H. 13. Jones S, Bernstein D, Bialer M, Dhawan A,
Structural bases of Wolman disease and cho- Hendriksz C, Whitley C, et al. Severe and ra-
lesteryl ester storage disease. Mol Genet pid disease course in the natural history of infants
Metab 2012;105:244-248. https://doi. with lysosomal acid lipase deficiency. Mol Ge-
org/10.1016/j.ymgme.2011.11.004. net Metab 2014;111:S57-S58. https://doi.
5. Goldstein JL, Dana SE, Faust JR, Beaudet AL, org/10.1016/j.ymgme.2013.12.125.
Brown MS. Role of lysosomal acid lipase in the 14. Beaudet AL, Ferry GD, Nichols BL, Jr., Rosen-
metabolism of plasma low density lipoprotein. berg HS. Cholesterol ester storage disease: cli-
Observations in cultured fibroblasts from a pa- nical, biochemical, and pathological studies.
tient with cholesteryl ester storage disease. J Biol J Pediatr 1977;90:910-914. https://doi.
Chem 1975;250:8487-8495. org/10.1016/s0022-3476(77)80557-x.
Volumen 3 | Número 1 | Ene-Jun 2022 103|Siuffi-Campo S, Londoño-García R, Espinosa-Herrera YP, Pérez-Cadavid JC, Muñoz-Maya OG
15. Zhang B, Porto AF. Cholesteryl ester storage 24. Scott SA, Liu B, Nazarenko I, Martis S, Kozlitina
disease: protean presentations of lysosomal J, Yang Y, et al. Frequency of the cholesteryl ester
acid lipase deficiency. J Pediatr Gastroente- storage disease common LIPA E8SJM mutation
rol Nutr 2013;56:682-685. https://doi. (c.894G>A) in various racial and ethnic groups.
org/10.1097/mpg.0b013e31828b36ac. Hepatology 2013;58:958-965. https://doi.
16. Drebber U, Andersen M, Kasper HU, Lohse P, org/10.1002/hep.26327.
Stolte M, Dienes HP. Severe chronic diarrhea 25. Chalasani N, Younossi Z, Lavine JE, Diehl AM,
and weight loss in cholesteryl ester storage Brunt EM, Cusi K, et al. The diagnosis and mana-
disease: a case report. World J Gastroen- gement of non-alcoholic fatty liver disease: practi-
terol 2005;11:2364-2366. https://doi. ce Guideline by the American Association for the
org/10.3748/wjg.v11.i15.2364. Study of Liver Diseases, American College of Gas-
17. Chora JR, Alves AC, Medeiros AM, Mariano troenterology, and the American Gastroenterolo-
C, Lobarinhas G, Guerra A, et al. Lysosomal gical Association. Hepatology 2012;55:2005-
acid lipase deficiency: A hidden disease among 2023. https://doi.org/10.1002/hep.25762.
cohorts of familial hypercholesterolemia? J Clin 26. Vajro P, Lenta S, Socha P, Dhawan A, Mc-
Lipidol 2017;11:477-484.e472. https://doi. Kiernan P, Baumann U, et al. Diagnosis of
org/10.1016/j.jacl.2016.11.002. nonalcoholic fatty liver disease in children and
18. Burton BK, Balwani M, Feillet F, Barić I, Burrow adolescents: position paper of the ESPGHAN
TA, Camarena Grande C, et al. A phase 3 trial Hepatology Committee. J Pediatr Gastroen-
of sebelipase alfa in lysosomal acid lipase defi- terol Nutr 2012;54:700-713. https://doi.
ciency. N Engl J Med 2015;373:1010-1020. org/10.1097/MPG.0b013e318252a13f.
https://doi.org/10.1056/NEJMoa1501365. 27. Hůlková H, Elleder M. Distinctive histopatholo-
19. Grabowski GA, Valayannopoulos V, Goodman gical features that support a diagnosis of cho-
ZD, Balwani M. Lysosomal acid lipase deficien- lesterol ester storage disease in liver biopsy
cy: The continuous spectra of disease variants. specimens. Histopathology 2012;60:1107-
The Online Metabolic and Molecular Bases of In- 1113. https://doi.org/10.1111/j.1365-
herited Disease. New York: McGraw-Hill; 2019. 2559.2011.04164.x.
20. Himes RW, Barlow SE, Bove K, Quintani- 28. Boldrini R, Devito R, Biselli R, Filocamo M, Bosman
lla NM, Sheridan R, Kohli R. Lysosomal acid C. Wolman disease and cholesteryl ester storage
lipase deficiency unmasked in two children disease diagnosed by histological and ultrastruc-
with nonalcoholic fatty liver disease. Pedia- tural examination of intestinal and liver biopsy.
trics 2016;138:e20160214. https://doi. Pathol Res Pract 2004;200:231-240. https://
org/10.1542/peds.2016-0214. doi.org/10.1016/j.prp.2003.11.001.
21. Riva S, Spada M, Sciveres M, Minervini M, Cin- 29. Thelwall PE, Smith FE, Leavitt MC, Canty D,
torino D, Maggiore G, et al. Hepatocarcinoma Hu W, Hollingsworth KG, et al. Hepatic cho-
in a child with cholesterol ester storage disea- lesteryl ester accumulation in lysosomal acid li-
se. Dig Liver Dis 2008;40:784. https://doi. pase deficiency: non-invasive identification and
org/10.1016/j.dld.2008.01.009. treatment monitoring by magnetic resonance.
22. Hamilton J, Jones I, Srivastava R, Galloway J Hepatol 2013;59:543-549. https://doi.
P. A new method for the measurement of ly- org/10.1016/j.jhep.2013.04.016.
sosomal acid lipase in dried blood spots 30. Reiner Ž. Statins in the primary preven-
using the inhibitor Lalistat 2. Clin Chim tion of cardiovascular disease. Nat Rev
Acta 2012;413:1207-1210. https://doi. Cardiol 2013;10:453-464. https://doi.
org/10.1016/j.cca.2012.03.019. org/10.1038/nrcardio.2013.80.
23. Mukherjee M. Human digestive and meta- 31. Xu M, Liu K, Swaroop M, Porter FD, Sidhu R,
bolic lipases-A brief review. J Mol Catal B Firnkes S, et al. δ-Tocopherol reduces lipid
Enzym 2003;22:369-376. https://doi. accumulation in Niemann-Pick type C1 and
org/10.1016/S1381-1177(03)00052-3. Wolman cholesterol storage disorders. J Biol
104 Deficiencia de lipasa ácida lisosomal, una enfermedad subdiagnosticada. Reporte de casoChem 2012;287:39349-39360. https://doi. J Hepatol 2014;61:1135-1142. https://doi.
org/10.1074/jbc.M112.357707. org/10.1016/j.jhep.2014.06.022.
32. Tolar J, Petryk A, Khan K, Bjoraker KJ, Jessurun 36. Wilson DP, Friedman M, Marulkar S, Hamby
J, Dolan M, et al. Long-term metabolic, endocri- T, Bruckert E. Sebelipase alfa improves athe-
ne, and neuropsychological outcome of hema- rogenic biomarkers in adults and children
topoietic cell transplantation for Wolman disea- with lysosomal acid lipase deficiency. J Clin
se. Bone Marrow Transplant 2009;43:21-27. Lipidol 2018;12:604-614. https://doi.
https://doi.org/10.1038/bmt.2008.273. org/10.1016/j.jacl.2018.02.020.
33. Stein J, Garty BZ, Dror Y, Fenig E, Zeigler M, Ya- 37. Pastores GM, Hughes DA. Lysosomal acid li-
niv I. Successful treatment of Wolman disease by pase deficiency: Therapeutic options. Drug Des
unrelated umbilical cord blood transplantation. Devel Ther 2020;14:591-601. https://doi.
Eur J Pediatr 2007;166:663-666. https://doi. org/10.2147/dddt.S149264.
org/10.1007/s00431-006-0298-6. 38. Camarena C, Aldamiz-Echevarria LJ, Polo
34. Balwani M, Breen C, Enns GM, Deegan PB, B, Barba Romero MA, García I, Cebolla
Honzík T, Jones S, et al. Clinical effect and JJ, et al. Actualización en deficiencia de li-
safety profile of recombinant human lysosomal pasa ácida lisosomal: diagnóstico, trata-
acid lipase in patients with cholesteryl ester sto- miento y seguimiento de los pacientes. Med
rage disease. Hepatology 2013;58:950-957. Clin 2017;148:e421-429. https://doi.
https://doi.org/10.1002/hep.26289. org/10.1016/j.medcli.2016.12.044.
35. Valayannopoulos V, Malinova V, Honzík T, Ba- 39. Gómez-Duarte C, García V, Botero V, Aristi-
lwani M, Breen C, Deegan PB, et al. Sebelipase zabal A, Echeverri G, Pachajoa H. Deficiencia
alfa over 52 weeks reduces serum transamina- de lipasa ácida lisosomal, una patología infre-
ses, liver volume and improves serum lipids in cuente. Gac Med Mex 2019;155:291-297.
patients with lysosomal acid lipase deficiency. https://doi.org/10.24875/gmm.18004024.
Volumen 3 | Número 1 | Ene-Jun 2022 105También puede leer

















































