Clasificación de las miocardiopatías. Un objetivo, muchas propuestas - SciELO
←
→
Transcripción del contenido de la página
Si su navegador no muestra la página correctamente, lea el contenido de la página a continuación

Artículo de revisión ESPECIAL
Rev Urug Cardiol 2019; 34: 99-113
doi: 10.29277/cardio.34.1.19 MIOCARDIOPATÍAS
Clasificación
Jorge Estigarribia
de lasPassaro
miocardiopatías. Un objetivo, muchas propuestas
Clasificación de las miocardiopatías.
Un objetivo, muchas propuestas
Dr. Jorge Estigarribia Passaro
Resumen
Resulta innegable la importancia de la patología del miocardio como causa de enfermedad y muerte de origen cardíaco.
Actualmente, cerca de la mitad de los pacientes que fallecen súbitamente en la niñez y la adolescencia o que reciben un
trasplante cardíaco están afectados por una miocardiopatía.
No obstante, las enfermedades del miocardio han constituido históricamente un grupo desconcertante, tanto en relación
con su origen como en su sistematización nosológica.
Identificadas inicialmente con patología inflamatoria y concebidas luego como desórdenes de causa desconocida o mani-
festación de múltiples enfermedades sistémicas, las miocardiopatías fueron más tarde categorizadas en ciertos patrones
de presentación morfológico-funcional y objeto en los últimos 30 años de intensa investigación en el ámbito de las cien-
cias básicas, que permitió reconocer el origen genético de muchas de estas entidades.
Esa nueva información sustentó, ya en este siglo, iniciativas de clasificación por parte de la American Heart Association
(AHA) y la European Society of Cardiology (ESC), que a pesar de representar un valioso avance con respecto a intentos pre-
vios, muestran áreas de incertidumbre y discrepancias sustantivas que son objeto de debate. Una propuesta más reciente, la
clasificación MOGE(S), pone énfasis en la creciente información aportada por la genética molecular y en la implementación
de una nosología descriptiva fenotipo-genotipo que posibilite la máxima precisión en la nomenclatura y el diagnóstico clínico.
En este capítulo se revisa la evolución de los conceptos que sustentaron las sucesivas clasificaciones publicadas y se ana-
lizan las diferencias entre las propuestas de la AHA y la ESC, concluyendo en la necesidad de un abordaje conjunto del
problema en pos de una nomenclatura y un ordenamiento taxonómico coherentes y universalmente compartidos.
Palabras clave: MIOCARDIOPATÍAS
CLASIFICACIÓN
CONTROVERSIA
Classification of cardiomyopathies. One goal, many proposals
Summary
The importance of myocardial pathology as a cause of illness and death from cardiac origin is undeniable. Currently, al-
most half of the patients who die suddenly in childhood and adolescence or receive a heart transplant are affected by car-
diomyopathy. However, myocardial diseases have historically constituted a perplexing group, both in relation to their
origin and in their nosological systematization.
Initially identified with inflammatory pathology, and then conceived as disorders of unknown cause or as a manifesta-
tion of multiple systemic diseases, cardiomyopathies were later categorized in certain patterns of morphological and
functional presentation, and were object in the last 30 years of intense research in the field of basic sciences, which allo-
wed to recognize the genetic origin of many of these entities.
Already in this century, that new information sustained classification initiatives by the American Heart Association
(AHA) and the European Society of Cardiology (ESC), which despite being a valuable improvement over previous attempts,
exhibit areas of uncertainty and substantive differences that are subject of debate. A more recent proposal, the MOGE (S)
classification, stresses on the growing information provided by molecular genetics and on the implementation of a phenoty-
pe-genotype descriptive nosology that enables maximum accuracy in nomenclature and clinical diagnosis.
This chapter reviews the evolution of the concepts that sustained the successive classifications published, and analyzes
the discrepancies between the proposals of the AHA and the ESC, concluding on the need for a joint approach to the pro-
blem in order to generate a coherent and universally shared taxonomic arrangement and nomenclature.
Key words: CARDIOMYOPATHIES
CLASSIFICATION
CONTROVERSY
Departamento de Cirugía. Instituto Nacional de Cirugía Cardíaca.
Correspondencia: Dr. Jorge Estigarribia. Luis A. de Herrera 2275. Montevideo, Uruguay.
Correo electrónico: jorgeestigarribia@movinet.com.uy
El autor declara no tener conflictos de intereses.
Recibido Mar 3, 2019; aceptado Mar 4, 2019
99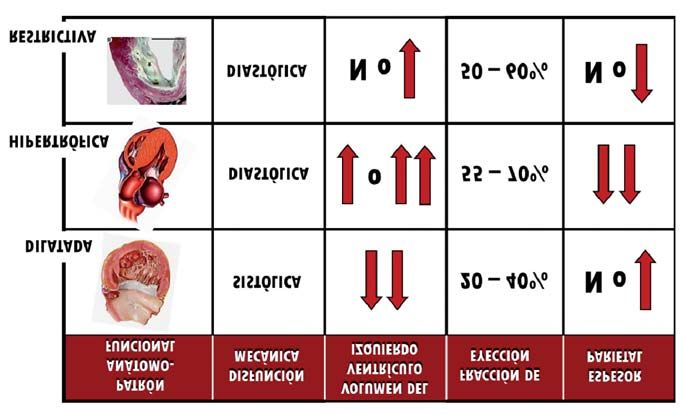
Clasificación de las miocardiopatías. Un objetivo, muchas propuestas Revista Uruguaya de Cardiología
Jorge Estigarribia Passaro Volumen 34 | nº 1 | Marzo 2019
Classificação das cardiomiopatias. Um objetivo, muitas propostas
Resumo
A importância da patologia miocárdica como causa de doença e morte de origem cardíaca é inegável. Atualmente, cerca
de metade dos pacientes que morrem subitamente na infância e adolescência ou que recebem um transplante cardíaco
são afetados pela cardiomiopatia.
No entanto, historicamente, as doenças do miocárdio têm sido um grupo desconcertante, tanto em relação à sua origem
quanto em sua sistematização nosológica.
Inicialmente identificadas com patologia inflamatória e, em seguida concebidas como doenças de causa desconhecida ou
manifestação de muitas doenças sistêmicas, as cardiomiopatias foram posteriormente categorizadas em certos padrões
de apresentação morfofuncional e objeto nos últimos 30 anos de intensa investigação no campo das ciências básicas, o
que permitiu reconhecer a origem genética de muitas dessas entidades.
Essa nova informação sustentou, já neste século, iniciativas de classificação da American Heart Association (AHA) e da
European Society of Cardiology (ESC), que apesar de representar um valioso avanço sobre as tentativas anteriores mos-
tram áreas de incerteza e discrepâncias substanciais que são objeto de debate. Uma proposta mais recente, a classificação
MOGE (S), enfatiza a crescente informação fornecida pela genética molecular e a aplicação de uma nosología descritiva
fenótipo-genótipo, permitindo a maior precisão no diagnóstico clínico e nomenclatura.
Neste capítulo é revista a evolução dos conceitos que sustentaram as classificações sucessivas publicadas, e as diferenças
entre as propostas da AHA e da ESC são analisadas, concluindo na necessidade de um abordagem conjunto do problema
em busca de uma nomenclatura e ordem taxonômica coerente e universalmente compartilhada.
Palavras-chave: CARDIOMIOPATIAS
CLASSIFICAÇÃO
CONTROVÉRSIA
miento relativo a su etiología, patogenia e historia
“No hay educación si no hay verdad que transmitir, si
todo es más o menos verdad, si cada cual tiene su natural, exceptuando quizás la frecuente compro-
verdad más o menos respetable y no se puede decidir bación de su incidencia familiar. Una muestra de tal
racionalmente entre tanta diversidad”. desorientación la constituyen los casi 60 términos
Fernando Savater, filósofo español. que se han utilizado para designar a una misma en-
tidad, hoy conocida como miocardiopatía hipertrófi-
ca(1,2) (MCH), o la denominación de la miocardiopa-
Introducción tía dilatada (MCD) con el nombre del país desde
Pocos capítulos de la cardiología han generado más donde procede el informe original o el de sus autores
dudas, confusiones y controversias que el de las en- principales.
fermedades del miocardio, y su nomenclatura y clasi- En época reciente, los notables avances en cien-
ficación han constituido un desafío ininterrumpido cias básicas (fundamentalmente la biología molecu-
hasta nuestros días. Un principio general para todos lar y la genética aplicada) han develado el origen ge-
los sistemas de clasificación de entidades nosológicas nético de muchas MP, otorgándoles finalmente
es su condicionamiento por el nivel de información identidad nosológica, y nuevas entidades han sido
disponible en la época en que se elaboran, y las mio- descritas. No obstante, a causa de su variadísima
cardiopatías (MP) son un ejemplo demostrativo. La etiología, su extraordinario grado de complejidad y
evolución de la información en este campo ha condu- su frecuente superposición genotípica y fenotípica,
cido a la publicación de múltiples clasificaciones su- persisten importantes incongruencias tanto con-
cesivas a cargo de investigadores individuales o de ceptuales como semánticas en relación con su no-
consensos de expertos que representaban la posición menclatura y ubicación taxonómica.
oficial de organizaciones médicas globales, como la En este capítulo nos proponemos revisar la evo-
World Health Organization (WHO) y la Internatio- lución de los conceptos que dieron origen a las prin-
nal Society and Federation of Cardiology (ISFC) o de cipales clasificaciones publicadas, para posterior-
sociedades científicas del ámbito de la cardiología de mente abordar el estado actual del tema, profundi-
particular influencia en ambos lados del Atlántico. zando en las diferencias y aspectos debatibles de las
Luego de una prolongada etapa en la que la pa- propuestas europea y norteamericana actualmente
tología propia del músculo cardíaco fue ignorada o en vigencia. Finalmente, nos introduciremos en la
atribuida rutinariamente a inflamación de causa más reciente clasificación MOGE(S), que capitali-
desconocida, siguió el reconocimiento de algunos zando los conocimientos adquiridos en las últimas
patrones anatomofuncionales particulares de afec- tres décadas y no obstante su relativa complejidad,
tación miocárdica, aunque con escaso o nulo conoci- representa un cambio de orientación desde el mero
100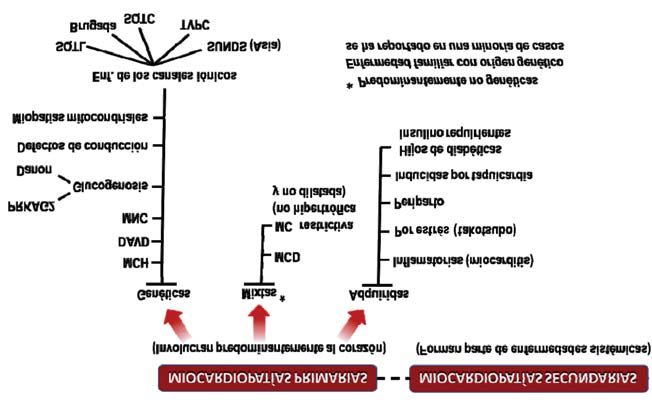
Revista Uruguaya de Cardiología Clasificación de las miocardiopatías. Un objetivo, muchas propuestas
Volumen 34 | nº 1 | Marzo 2019 Jorge Estigarribia Passaro
Figura 1. La primera clasificación en tres tipos básicos de comportamiento clínico de las miocardiopatías corresponde a J.
F. Goodwin y colaboradores, en un artículo del cual se reproduce aquí el encabezamiento junto con la fotografía del autor
principal. Goodwin utilizó un criterio fisiopatológico en base al cual dividió las miocardiopatías en congestiva, constricti-
va (u obliterativa) y obstructiva, terminología que posteriormente fue modificada con la introducción de elementos mor-
fológicos, lo que no menoscaba el valor clínico de su concepción original.
ordenamiento taxonómico hacia la aplicación prác- va(6), con lo que resurgió para este grupo de causa
tica en el diagnóstico clínico y la descripción detalla- incierta la expresión “enfermedad miocárdica pri-
da de la enfermedad en el paciente individual. maria”(7) .
No es objetivo de esta revisión la descripción in- En 1956, Blankenhorn y Gall usaron el término
dividual de las distintas MP, que han sido bien ca- miocarditis para designar la patología inflamatoria
racterizadas en numerosas publicaciones dedicadas del músculo cardíaco, y miocardosis para otro tipo
a las entidades específicas. de afecciones consideradas “degenerativas”(8).
El término cardiomiopatía fue utilizado por pri-
mera vez por W. Brigden en 1957(9) para describir la
La patología miocárdica en retrospectiva enfermedad miocárdica de origen no coronario* y la
Durante la segunda mitad del siglo XIX se mencio- clasificó en cinco formas según su origen: congéni-
naba la existencia de la miocarditis crónica, conce- ta, infecciosa, por enfermedad del colágeno, amiloi-
bida entonces como cualquier enfermedad del dosis, y una variedad anatómica, la fibrosis endo-
músculo cardíaco de origen no valvular y única pa- miocárdica, de etiología incierta. Insistió en restrin-
tología propia del miocardio reconocida hasta fines gir el nombre de miocarditis específicamente a la
del siglo, cuando comenzó a hablarse de enfermeda- patología del miocardio de origen infeccioso y previó
des del músculo cardíaco con una perspectiva más la progresiva reducción del grupo de las MP llama-
amplia. das idiopáticas a medida que diversos agentes infec-
En Alemania, Krehl introdujo el concepto de en- ciosos causales fueran identificados.
fermedad idiopática del músculo cardíaco en 1891(3), Con frecuencia se cita la publicación de Good-
mientras que Josserand y Gallavardin en Francia win y Oakley de 1972(12) como la primera clasifica-
utilizaron el término enfermedad miocárdica pri- ción en los tres tipos morfológico-funcionales que
maria en 1901 para la afectación miocárdica de cau- hasta hoy utilizamos para el manejo clínico de las
sa desconocida, visualizando sin embargo la posibi- MP. Sin embargo, esta concepción reconoce un an-
lidad de otras etiologías(4). tecedente en el trabajo que el propio J. F. Goodwin
Warren, en 1933, señaló lo inusual de la confir- (figura 1), junto con otros autores, publicó en 1961(13),
mación diagnóstica de miocarditis crónica en la au-
topsia(5), con lo que la atención se centró en etiolo- * Si bien la angina de pecho fue descrita por Heberden en
gías no inflamatorias, en especial la enfermedad co- 1768, los diferentes correlatos clínicos de la oclusión co-
ronaria (EC) y la hipertensión arterial (HTA). ronaria comenzaron a conocerse mejor desde 1912 con
Advirtió dos modalidades de muerte en las MP: por las publicaciones de James Herrick(10,11), por lo cual la
deterioro gradual de una insuficiencia cardiorrespi- preocupación de los clínicos al momento de diagnosticar
ratoria o de forma súbita. una enfermedad propia del miocardio consistía en el si-
En 1950, H. Christian destacó que un tercio de glo XIX en descartar la patología valvular, conocida des-
la enfermedad no inflamatoria cardíaca no puede de antaño, y recién bien entrado el siglo XX involucró a
adjudicarse a enfermedad coronaria o hipertensi- la EC, de prevalencia y reconocimiento crecientes.
101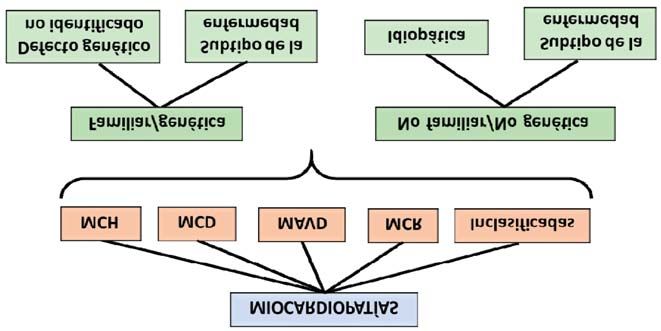
Clasificación de las miocardiopatías. Un objetivo, muchas propuestas Revista Uruguaya de Cardiología
Jorge Estigarribia Passaro Volumen 34 | nº 1 | Marzo 2019
Figura 2. Modelo de clasificación de las miocardiopatías según su fenotipo estructural y funcional, adoptado en el primer
esfuerzo global de sistematización nosológica de las miocardiopatías a cargo de un Comité de la WHO/ISFC en 1980, cri-
terio que la ESC mantendrá en su clasificación de 2008. Posteriormente al documento de 1980, se incorporarán otras en-
tidades que no tienen cabida en este esquema, como la miocardiopatía arritmogénica del ventrículo derecho, la miocar-
diopatía no compactada y el síndrome de takotsubo. N: normal.
WHO: World Health Organization; ISFC: International Society and Federation of Cardiology; ESC: European Society
of Cardiology
donde definió las miocardiopatías como “enfermeda- Esfuerzos institucionales de clasificación
des del músculo cardíaco subagudas o crónicas de de las miocardiopatías
etiología oscura o desconocida”, y distinguió por
primera vez tres formas de presentación clínica que Clasificación WHO/ISFC 1980(16)
denominó: congestiva (posteriormente dilatada),
Se trata de una publicación breve y con algunas in-
obliterativa o constrictiva (por su similitud fisiopa-
consistencias.
tológica con la pericarditis constrictiva y ahora co-
El documento define las MP como “enfermeda-
nocida como restrictiva), y obstructiva (actualmen-
des del músculo cardíaco de causa desconocida”, di-
te hipertrófica), división que hasta hoy continúa
vidiéndolas, siguiendo a Goodwin, en tres patrones
siendo de utilidad metodológica para el diagnóstico
básicos: dilatada, hipertrófica y restrictiva (MCR),
clínico.
con una mínima caracterización de cada una. La fi-
Fowler, en 1964, publicó su clasificación(14), en gura 2 muestra las características anatomofuncio-
la cual denominó como “primarias” a las MP idiopá- nales básicas que permiten diferenciar dichos pa-
ticas, advirtiendo que en el futuro podrían identifi- trones.
carse dentro de ellas varias categorías etiológicas, Describe, diferenciándolo de las MP, un amplio
de las cuales la miocarditis podría ser una causa grupo de enfermedades del miocardio causadas por
muy frecuente. o asociadas a patologías de otros sistemas, a las que
Un boletín de la WHO de 1968(15) también desig- denomina “enfermedades específicas del músculo
nó como primarias a las MP de etiología desconoci- cardíaco”, excluyendo expresamente la afectación
da y secundarias a las asociadas a una enfermedad del miocardio originada en la HTA sistémica o pul-
sistémica o a un síndrome conocidos. monar, la EC, las valvulopatías (VP) y las cardiopa-
Ante este confuso panorama, diversos comités tías congénitas (CC).
de expertos designados por instituciones mundiales Estas enfermedades miocárdicas específicas –no
de la salud o sociedades científicas de la especialidad reconocidas aquí como MP– son clasificadas según
a lo largo de las décadas siguientes intentaron orde- su causa en infecciosas, metabólicas, heredofamilia-
nar la información disponible y generaron docu- res, por toxicidad e hipersensibilidad, y por patolo-
mentos con su visión del problema según el estado gías sistémicas.
del conocimiento en cada época. Ello no impidió, sin La diferenciación, sin embargo, parece más se-
embargo, que persistieran puntos oscuros, contra- mántica que de fondo, considerando que “miocar-
dicciones y diferencias conceptuales que señalare- diopatía” no significa otra cosa que “enfermedad
mos en esta revisión. del músculo cardíaco” sea idiopática o no, y que no
102Revista Uruguaya de Cardiología Clasificación de las miocardiopatías. Un objetivo, muchas propuestas
Volumen 34 | nº 1 | Marzo 2019 Jorge Estigarribia Passaro
resulta clara la razón por la que un desorden del Señala que la MCD puede ser tanto idiopática
músculo cardíaco originado en una patología sisté- como de etiología familiar/genética, viral o inmune,
mica no debiera considerarse como MP específica o ambas, o tóxica/alcohólica, y que la MCH, de pre-
una vez excluidas la HTA, la EC y las VP. Este crite- sentación familiar con herencia autosómica domi-
rio sería reconsiderado en una versión posterior en nante, es causada por mutaciones en genes de pro-
1995. teínas contráctiles del sarcómero.
Esta clasificación de la WHO precedió al notable Acepta que algunas arritmias y trastornos de la
desarrollo de la genética aplicada a las enfermeda- conducción pueden constituir enfermedades mio-
des cardiovasculares que en las últimas tres déca- cárdicas primarias, pese a lo cual el panel de exper-
das logró identificar los genes responsables de mu- tos elige no incluirlas entre las MP.
chas MP(17-19), por lo cual su asociación con enferme- Razonablemente, sustituye la denominación
dad idiopática es compartible. Sin embargo, en el “Enfermedades específicas del músculo cardíaco”
caso específico de la MCH el documento reconoce un de la versión previa por “Miocardiopatías específi-
patrón de herencia monogénico autosómico domi- cas”, asociadas con desórdenes sistémicos particu-
nante, que conlleva implícito su origen genético lares que categoriza en etiologías inflamatoria, me-
–aunque sin identificación aún de los genes causa- tabólica, inmunológica, miopática, neuromuscular,
les–, por lo que no sería adecuada, incluso en aquel tóxica y periparto, incluyendo la mayoría de ellas
contexto temporal, la definición general de MP co- varias enfermedades causales.
mo desorden de origen desconocido. De hecho, la de- Sorprendentemente, incluye también en este
signación que emplea “miocardiopatía hipertrófi- grupo de verdaderas MP la afectación miocárdica
ca”, utilizada en la actualidad, evita el adjetivo asociada con EC, HTA y VP, denominándolas “MP
“idiopática” incluido en gran parte de la nomencla- isquémica, hipertensiva y valvular”, respectiva-
tura que le fue aplicada previamente. Esta incon- mente, descartadas tanto históricamente como en
gruencia probablemente se deba a una inercia his- la versión anterior de la WHO por entender que
tórica del concepto por el cual una MP que no forma “ampliar de esta forma el panorama etiológico po-
parte de una enfermedad identificable era conside- dría tornar inútil la clasificación”. La condición que
rada idiopática o primaria. Como veremos luego, el panel de expertos requirió para fundamentar tal
las acepciones de los términos “primaria” y “secun- viraje conceptual fue que el deterioro de la función
daria” han variado en paralelo al desarrollo de la contráctil del ventrículo no pueda explicarse por la
biología molecular, que ha permitido desconectar el extensión de la EC o del daño isquémico, o resulte
concepto de MP con el de enfermedad de causa desproporcionado con las condiciones anormales de
desconocida. carga. Esta explicación resulta poco consistente, ya
Este documento agrega además un oscuro grupo que si se admite que el daño miocárdico no puede
de “Miocardiopatías inclasificadas”, que no tiene adjudicarse a la enfermedad de fondo, debería con-
cabida en ninguno de los previos, y que lo concibe siderarse como una segunda enfermedad de causa
como enfermedades con alteraciones menores que no determinada y no sería clasificable como una MP
podrían evolucionar o no hacia la MP manifiesta, específica sino idiopática, si no hay otra causa detec-
por lo que también las han denominado “Miocar- table.
diopatías latentes”. Como en el documento previo, continúa exis-
tiendo un subgrupo de MP no clasificadas con mejor
Clasificación WHO/ISFC 1995-1996(20) identificación de sus integrantes, que incluyen la fi-
Esta versión define las MP como “enfermedades del broelastosis, la miocardiopatía no compactada
miocardio asociadas con disfunción cardíaca”. (MNC) descrita en 1990(22), la disfunción miocárdi-
Abandona la distinción entre MP (entendidas en la ca con dilatación mínima y las MP de origen mito-
versión previa como idiopáticas) y enfermedades es- condrial.
pecíficas del músculo cardíaco, ya que la confirma-
ción de causas tanto genéticas como adquiridas en Clasificación de la American Heart Association
los tres tipos morfofuncionales básicos eliminó la 2006(23)
condición que supuestamente las diferenciaba. Ya Comienza señalando las históricas confusiones y
no era aceptable que el diagnóstico de MP se contradicciones en la nomenclatura y clasificación
condicionara al desconocimiento de su etiología. de las MP, que entiende han menoscabado su com-
Además de la MCD, la MCH y la MCR, incluye la prensión y su apropiada consideración en la clínica.
miocardiopatía arritmogénica del ventrículo dere- Aboga por mejorar la precisión del lenguaje emplea-
cho (MAVD), inicialmente denominada displasia do y expone la necesidad de incorporar los avances
arritmogénica del VD y caracterizada en 1982(21). de la genética molecular en el diagnóstico etiológico
103Clasificación de las miocardiopatías. Un objetivo, muchas propuestas Revista Uruguaya de Cardiología
Jorge Estigarribia Passaro Volumen 34 | nº 1 | Marzo 2019
y las nuevas entidades descritas en la última déca- De esta definición y del desarrollo del documen-
da, incluyendo las canalopatías iónicas con su po- to surgen varios puntos destacables; varios de ellos
tencial arritmogénico letal, y considera obsoleta en novedosos con respecto a clasificaciones previas:
varios aspectos la clasificación de la WHO de 1995.
a) No es necesario que exista disfunción miocárdi-
Propone una clasificación que considera en ne- ca mecánica sistólica o diastólica ni un correlato
cesaria correspondencia con la era molecular de las morfológico identificable en los estudios de ima-
cardiopatías y con aplicación en el diagnóstico clíni- gen, biopsia o necropsia para considerar el diag-
co, sin pretender proporcionar una metodología o nóstico de MP. Por lo tanto, la AHA incluye afec-
estrategia específica para este fin, admitiendo que ciones puramente eléctricas caracterizadas por
requerirá revisión en el futuro. un elevado riesgo de arritmias letales cuyas ano-
Presenta una revisión crítica de la clasificación malías funcionales y estructurales se encuen-
en MCD, MCH y MCR, en base a las siguientes con- tran a nivel molecular, como es el caso de las ca-
sideraciones: nalopatías iónicas. Estas enfermedades no se
detectan en un estudio de imagen, sino que se
1. Mezcla criterios anatómicos (dilatación, hiper-
manifiestan en el electrocardiograma (ECG).
trofia) con uno funcional (restricción), con lo
b) En oposición a la segunda clasificación de la
cual la misma enfermedad podría ubicarse en
WHO, reafirma la clásica exclusión de las afec-
dos categorías (por ejemplo, la MCH exhibe tan-
ciones del miocardio producto de HTA, EC, VP y
to hipertrofia como restricción).
CC, descartando, por ejemplo, la frecuente ex-
2. Algunas formas de MP pueden no tener un feno- presión “miocardiopatía isquémica”.
tipo estático y sufrir remodelación, pasando de c) Establece una división inicial entre MP prima-
una categoría a otra, como la MCH o la cardiopa- rias y secundarias, aunque con un significado
tía amiloidea, que eventualmente evolucionan a diferente del etiológico histórico, definiendo con
una MCD. un criterio de contexto clínico como primarias
3. La identificación del sustrato genético de la en- aquellas MP que se encuentran única o predo-
fermedad puede preceder en años a cualquier al- minantemente confinadas al corazón, en núme-
teración morfológica o funcional objetivable, ro relativamente limitado, y como secundarias
con lo cual no aplican aún las características las que forman parte de una gran variedad de
cuestionadas. enfermedades sistémicas o multiorgánicas.
4. En ciertas oportunidades resulta difícil distin- Éstas habían sido denominadas “Enfermedades
guir formas dilatadas de no dilatadas en base a específicas del músculo cardíaco” o “Miocardio-
medidas cuantitativas, dado que el diámetro patías específicas” por la WHO en 1980 y 1995,
ventricular es una magnitud continua y hay ex- respectivamente.
presiones incipientes de la enfermedad. El documento reconoce que ocasionalmente es-
ta distinción puede ser arbitraria, dado que al-
Concluye, en definitiva, que la clasificación mor- gunas MP pueden afectar predominantemente
fofuncional se ha tornado de poca utilidad y que pro- pero no de forma exclusiva al corazón, y la deci-
bablemente debería ser abandonada. sión recae en la valoración de la importancia clí-
También expone las limitaciones propias de las nica y las consecuencias del proceso miocárdico.
clasificaciones puramente etiológicas, donde un d) A su vez, subclasifica las MP primarias según la
mismo patrón anatomofuncional puede responder a naturaleza de su origen en genéticas, mixtas (ge-
múltiples etiologías, y las funcionales, dado que este néticas y no genéticas) y adquiridas (figura 3) y
enfoque tiene una aplicación fundamentalmente te- a continuación realiza una descripción de las ca-
rapéutica, pero las estrategias de tratamiento cam- racterísticas básicas de cada una. De esta des-
bian continuamente. cripción, un par de aspectos puntuales resultan
La AHA define las MP como “un grupo heterogé- relevantes:
neo de enfermedades del miocardio asociadas a dis- • La MCH es descrita como una MP primaria
función eléctrica y/o mecánica que usualmente (pero genética (afectando única o principalmente al
no invariablemente) exhiben inapropiada dilata- corazón) originada en una diversidad de mu-
ción o hipertrofia ventricular y son debidas a una di- taciones de proteínas del sarcómero codifica-
versidad de causas que frecuentemente son genéti- das por 11 genes. La distingue de una serie de
cas. Pueden estar confinadas al corazón o ser parte MP metabólicas, infiltrativas o por acumula-
de desórdenes sistémicos generalizados, y a menudo ción de productos, neurodegenerativas o mul-
conducen a muerte cardiovascular o incapacidad tiorgánicas (por lo tanto, secundarias) que se
progresiva relacionada con insuficiencia cardíaca”. deben a mutaciones en genes distintos a los
104Revista Uruguaya de Cardiología Clasificación de las miocardiopatías. Un objetivo, muchas propuestas
Volumen 34 | nº 1 | Marzo 2019 Jorge Estigarribia Passaro
Figura 3. Clasificación de las miocardiopatías según la American Heart Association, 2006.
MCH: miocardiopatía hipertrófica; DAVD: displasia (miocardiopatía) arritmogénica del ventrículo derecho; MNC: mio-
cardiopatía no compactada; PRKAG2: subunidad gamma-2 regulatoria de la proteinquinasa activada por el AMP; MCD:
miocardiopatía dilatada; SQTL: síndrome de QT largo; SQTC: síndrome de QT corto; TVPC: taquicardia ventricular po-
limórfica catecolaminérgica; SUNDS: síndrome de muerte súbita inesperada nocturna.
que codifican las proteínas sarcoméricas. Esta También desestima la posibilidad de una clasifi-
posición es refrendada por la Guía para el cación esencialmente genético-etiológica, argumen-
Diagnóstico y Tratamiento de la MCH publi- tando que “en la práctica clínica el camino desde el
cada por la American College of Cardiology diagnóstico al tratamiento no comienza en general
Foundation (ACCF), junto a la AHA, cinco con el hallazgo de una mutación”, y que “en caso de
años después de este documento(24). identificación de un gen patogénico a nivel familiar
• Como mencionáramos previamente, esta cla- la comprobación de una enfermedad clínicamente
sificación considera como MP a las canalopa- relevante en un portador requiere demostrar un fe-
tías iónicas, incluyendo los síndromes de QT notipo morfológico”.
largo y QT corto, el síndrome de Brugada y la De todas formas, llega al mismo punto al dividir-
taquicardia ventricular polimórfica catecola- las en genéticas/familiares y no genéticas/no fami-
minérgica. También se mencionan el síndro- liares, aunque en su algoritmo de clasificación ante-
me de muerte súbita inesperada nocturna, ponga el criterio morfológico.
que es el equivalente asiático del síndrome de La ESC considera que “una miocardiopatía es
Brugada en su base clínica y genética, y la un desorden en el cual el músculo cardíaco es estruc-
enfermedad de Lenegre del sistema de con- tural y funcionalmente anormal, en ausencia de en-
ducción. fermedad coronaria, hipertensión arterial, enferme-
dad valvular y enfermedad cardíaca congénita sufi-
ciente para causar la anormalidad miocárdica ob-
Clasificación de la European Society of servada”.
Cardiology 2008(25) Como se desprende de la definición, la ESC dis-
Señala la escasez de conocimientos sobre la etiolo- crepa con la AHA en relación con las canalopatías,
gía y la fisiopatología de las MP que caracterizó a las argumentando contra su interpretación como ver-
clasificaciones previas, aun comprendiendo que in- daderas MP, ya que requiere una alteración estruc-
volucraban entidades diferentes. tural objetivable en los estudios de imagen.
Reconoce que la distinción entre MP primarias Las clasifica en fenotipos morfológico-funciona-
y secundarias, según la acepción clásica por la cual les y cada fenotipo es subclasificado en formas fami-
primaria se asimilaba a idiopática, ha perdido vi- liares y no familiares (figura 4), entendiendo por fa-
gencia al develarse la etiología genética de muchas miliar o genética la ocurrencia en más de un miem-
MP antes denominadas primarias, y renuncia a esa bro de la familia de la misma enfermedad o de un fe-
terminología, realizando sin embargo un comenta- notipo que pueda ser causado por la misma muta-
rio crítico sobre el significado diferente de esos tér- ción y no por enfermedades cardíacas o sistémicas
minos que utilizó la AHA en su documento. en las que el fenotipo clínico sea influenciado por
105Clasificación de las miocardiopatías. Un objetivo, muchas propuestas Revista Uruguaya de Cardiología
Jorge Estigarribia Passaro Volumen 34 | nº 1 | Marzo 2019
Figura 4. Clasificación de las miocardiopatías según la European Society of Cardiology, 2008. MCH: miocardiopatía hi-
pertrófica; MCD: miocardiopatía dilatada; MAVD: miocardiopatía arritmogénica del ventrículo derecho; MCR: miocar-
diopatía restrictiva.
polimorfismo genético, dado que la mayoría de las res, en la cual se expresan con letras mayúsculas las
MP familiares son monogénicas. categorías relevantes que representan la extensión
Las MP no familiares son subdivididas en idio- de la enfermedad (Tumor primario-Nódulo linfáti-
páticas si no tienen causa identificable, y adquiri- co-Metástasis), y se define con un subíndice inte-
das si la disfunción ventricular es una complicación grado por números y letras la situación particular
y no un rasgo propio de la enfermedad. del paciente en cada categoría.
El documento afirma erróneamente que en la En esencia, MOGE(S) no representa una verda-
clasificación de la WHO/ISFC de 1995 se excluyó la dera clasificación, concebida como un cuadro teóri-
disfunción ventricular producto de EC, HTA, VP o co de entidades nosológicas agrupadas en categorías
CC. Aunque estas etiologías fueron excluidas en la como los que describimos previamente, sino un sis-
clasificación de 1980, en el documento de 1995 fue- tema descriptivo de nomenclatura y notación apli-
ron incorporadas dentro del concepto de MP especí- cado al diagnóstico en la práctica clínica, basado en
ficas, introduciendo un requisito diagnóstico poco un conocimiento más profundo de las bases fisiopa-
compartible, previamente explicado. En la clasifica- tológicas y genéticas de las MP. Su objetivo es preci-
ción de la ESC estas condiciones clínicas quedan sar todos los atributos clínicos y genéticos que pre-
claramente descartadas del grupo de las MP, pero senta una determinada MP en el paciente indivi-
se reedita la expresión “enfermedades específicas dual (fenotipo-genotipo) a fin de proveer un diag-
del músculo cardíaco” (empleada en la clasificación nóstico descriptivo detallado para su manejo, gene-
de la WHO de 1980 con otro significado) para ubi- rando a la vez una base común con un propósito de
carlas. investigación.
La tabla 1 sintetiza las principales diferencias Los proponentes consideran que la clasificación
conceptuales entre ambas clasificaciones. morfológico-funcional continúa teniendo relevan-
cia, pero la evaluación basada en el genotipo debe
Clasificación MOGE(S) 2013(26,27) ser la que dirija el proceso diagnóstico y las decisio-
Tomando nota de las limitaciones y puntos contro- nes terapéuticas en el paciente afectado y sus fami-
versiales de los sistemas de clasificación preceden- liares, así como las estrategias de seguimiento.
tes, y sustentándose en una caracterización no in- En su publicación original, los autores definen a
vasiva más refinada del fenotipo y el creciente cono- las MP como “desórdenes caracterizados por un
cimiento de la etiología genética precisa de muchas miocardio morfológica y funcionalmente anormal
MP, surgió una iniciativa de ordenamiento nosoló- en ausencia de cualquier otra enfermedad que sea
gico con un enfoque esencialmente distinto, en cuya suficiente por sí misma para causar el fenotipo ob-
concepción participaron cardiólogos clínicos, espe- servado”.
cialistas en insuficiencia cardíaca y trasplante, Esta definición resulta algo desconcertante,
genetistas e imagenólogos tanto europeos como es- porque a diferencia de las publicaciones de la ESC y
tadounidenses. la AHA, que descartan explícitamente la EC, la VP,
Esta propuesta, avalada por el Comité Científico la HTA y la CC, con la expresión “cualquier otra en-
de la World Heart Federation (WHF), está inspira- fermedad” parecería que solo incluyera las MP de-
da en el sistema de estadificación TNM de los tumo- nominadas primarias por la AHA. Por ejemplo, la
106Revista Uruguaya de Cardiología Clasificación de las miocardiopatías. Un objetivo, muchas propuestas
Volumen 34 | nº 1 | Marzo 2019 Jorge Estigarribia Passaro
Tabla 1. Diferencias entre las clasificaciones de la AHA y de la ESC.
AHA 2006 ESC 2008
Criterio de clasificación primariamente Criterio de clasificación primariamente morfológico.
etiológico-genético. División secundaria en genéticas/no genéticas.
En las miocardiopatías la disfunción miocárdica puede Requiere alteraciones estructurales objetivables en
ser estructural o exclusivamente eléctrica. Incluye las estudios de imagen macro o microscópica. Excluye las
canalopatías. canalopatías.
Todas las miocardiopatías están incluidas en algún Contempla un grupo de “miocardiopatías
grupo de la clasificación. inclasificadas” (takotsubo, miocardio no compactado).
Adopta la división en miocardiopatías primarias y Abandona la clasificación en primarias y secundarias,
secundarias con un criterio de contexto clínico concebida históricamente con un criterio etiológico
(afectación cardíaca aislada o multiorgánica). (primaria = idiopática).
Como ha sido la posición histórica, descarta la HTA, la También descarta esas etiologías, pero retoma la
EC, las VP y las CC como causas de miocardiopatía. confusa expresión “enfermedades específicas del
Deplora el término “miocardiopatía isquémica”. músculo cardíaco” para designar la afectación
miocárdica por estas causas.
Define la MCH como una hipertrofia parietal no Establece el diagnóstico de MCH solamente en base a
explicable por condiciones de carga, debida a criterios morfológicos. Incluye mutaciones no
mutaciones en proteínas contráctiles del sarcómero. sarcoméricas (fenocopias) exceptuando la cardiopatía
amiloidea.
Clasifica la miocarditis como una miocardiopatía Clasifica la miocarditis como una miocardiopatía no
primaria adquirida de naturaleza inflamatoria. familiar/no genética (secundaria) y le asigna una
ubicación morfológica dudosa dentro de las MCD.
AHA: American Heart Association; ESC: European Society of Cardiology; HTA: hipertensión arterial; EC: enfermedad coronaria;
VP: valvulopatías; CC: cardiopatías congénitas; MCH: miocardiopatía hipertrófica; MCD: miocardiopatías dilatadas.
cardiopatía amiloidea no sería, según esta defini- O: Compromiso de Órgano(s), que puede incluir
ción, una MP, ya que puede originar por sí misma solo al corazón (OH) o a muchos otros órganos o sis-
un fenotipo de hipertrofia, restricción o eventual- temas como músculo esquelético, (OH+M), riñón
mente dilatación. (OH+K), sistema nervioso, (OH+N), hígado (OH+L),
Dadas la complejidad clínica y etiológica de las etcétera, u O0 en caso de portadores sanos, dado
MP y su intención de realizar una caracterización que el corazón no está aún afectado.
exhaustiva, resultaría muy complejo brindar una G: Herencia Genética o familiar, precisando si
descripción completa y detallada de esta nomencla- es autosómica dominante (GAD), autosómica rece-
tura en la presente revisión, para cuya comprensión siva (GAR), ligada al cromosoma X (GXL), ligada al
cabal se remite al lector a las publicaciones origina- X recesiva (GXLR), ligada al X dominante (GXLD), o
les. transmisión matrilineal (GM). Se han previsto
Básicamente se ocupa de definir cinco atributos otros subíndices para casos esporádicos o con his-
de la MP en cuestión, donde las letras del acrónimo toria familiar negativa, desconocida o no investiga-
y los subíndices en idioma inglés tienen los siguien- da.
tes significados: E: Definición Etiológica. Resulta la notación más
M: Características Morfofuncionales que des- compleja, que refiere a la causa precisa de la enferme-
criben el fenotipo, con los subíndices MD por dilata- dad, sea genética o no. Si es genética, se designa EG
da, MH por hipertrófica, MA por arritmogénica del más el gen específico con la mutación sufrida. En
VD, MR por restrictiva y MLVNC por no compacta- el caso de una MCH, un ejemplo podría ser:
ción del VI. EG-MYH7(p.Arg403Glu). Puede tratarse de un no portador
Se prevén además múltiples posibilidades, como (EG-Neg) y hay múltiples opciones para designar pre-
superposiciones de fenotipos, atributos específicos sencia de más de una mutación o defectos genéticos
como bloqueo auriculoventricular, síndrome de complejos (EG-C), portadores obligados, no portadores
Wolf-Parkinson-White, onda épsilon, adquisición obligados, test genético no disponible o en curso, pa-
fenotípica temprana, ausencia de afectación en por- cientes genéticamente huérfanos, etcétera.
tador genético, etcétera, todas señaladas con sus En caso de etiología no genética puede describir-
respectivos subíndices. se como viral (V) agregando el virus, por ejemplo
107Clasificación de las miocardiopatías. Un objetivo, muchas propuestas Revista Uruguaya de Cardiología
Jorge Estigarribia Passaro Volumen 34 | nº 1 | Marzo 2019
Tabla 2. Evolución histórica de las propuestas de clasificación de las miocardiopatías durante los últimos 60
años (modificado de Arbustini et al [27]).
Año Definición / Clasificación Referencias
1956 Las enfermedades miocárdicas son clasificadas como miocarditis Blankenhorn y Gall(8)
(patología inflamatoria) o miocardosis (todas las demás causas,
concebidas esencialmente como patología “degenerativa”).
1957 Se propone el término miocardiopatía para designar la enfermedad Bridgen(9)
del miocardio poco frecuente de origen no coronario y se enfatiza en
la causa infecciosa de la miocarditis.
1961 Se describen las miocardiopatías como “enfermedades miocárdicas Goodwin, Gordon et al(13)
agudas o subagudas de etiología oscura o desconocida”,
clasificándose por primera vez en congestiva (hoy dilatada),
constrictiva (restrictiva) y obstructiva, denominada hipertrófica en
una versión posterior del mismo autor en 1972.
1980 Adopta la clasificación de Goodwin en tres tipos Informe de un Comité de Expertos
morfológico-funcionales. Se definen las miocardiopatías como de la WHO/ISFC sobre la
“enfermedades miocárdicas de etiología desconocida”. Se agrega el Definición y Clasificación de las
grupo de “enfermedades específicas del músculo cardíaco”, que tienen Miocardiopatías(16)
causa conocida y no se consideraron como miocardiopatías, y el de las
“miocardiopatías inclasificadas o latentes”.
1995-96 Actualización de la WHO/ISFC que define las miocardiopatías como Richardson et al. Segundo Informe
“enfermedades del miocardio asociadas con disfunción miocárdica”. de la WHO/ISFC(20)
Agrega la miocardiopatía arritmogénica del ventrículo derecho y
cambia la denominación “enfermedades específicas del músculo
cardíaco” por el de “miocardiopatías específicas” para la afectación
miocárdica de causa conocida.
1998 La ISFC se convierte en la WHF.
2006 La AHA define las miocardiopatías como “enfermedades del Clasificación AHA
miocardio asociadas con disfunción mecánica y/o eléctrica, que Maron et al(23)
usualmente (pero no invariablemente) exhiben hipertrofia o
dilatación ventricular inapropiada debido a una variedad de causas
que frecuentemente son genéticas”. Las clasifica en primarias y
secundarias según un criterio de contexto clínico. Representa el
primer intento visionario de clasificar las miocardiopatías primarias
priorizando los avances de la biología molecular y la genética
aplicada.
2008 La ESC define las miocardiopatías como “desórdenes del miocardio Clasificación ESC
en los cuales el músculo cardíaco es estructural y funcionalmente Elliot et al(25)
anormal”. Adopta la división en dilatada, hipertrófica, restrictiva,
arritmogénica del VD e inclasificadas, y las subclasifica en familiar
/genética y no familiar/no genética. Mantiene la vigencia del fenotipo
precediendo a la clasificación genética para el manejo en la práctica
clínica.
2013 La nosología WHF – MOGE(S) propone un sistema de nomenclatura Arbustini et al(26)
y notación descriptiva fenotipo-genotipo con propósitos de
diagnóstico clínico preciso e investigación.
WHO: World Health Organization; ISFC: International Society and Federation of Cardiology; WHF: World Heart Federation;
AHA: American Heart Association; ESC: European Society of Cardiology.
Coxsackie B3 (CB3) como EV-CB3 o Epstein-Barr vi- cluidas de las MP o por el reconocimiento de nuevas
rus (EBV) como EV-EBV. entidades.
Infecciones no virales se designan como EI más (S): Estadio (Stage) de insuficiencia cardíaca y
el agente específico si es posible, miocarditis como clase funcional.
EM. Hay también expresiones abreviadas para en- Se plantea como una definición opcional (de ahí
fermedades autoinmunes, amiloidosis no genética, el paréntesis), pero conveniente en particular para
sarcoidosis, causas tóxicas o por drogas, feocromoci- la descripción de una MP en etapa temprana. Se de-
toma, etcétera. finen entonces el estadio A a D de insuficiencia car-
La notación de la categoría E podrá modificarse díaca según la clasificación de la ACCF/AHA y la
en el futuro a causa de algunas condiciones hoy ex- clase funcional (CF) I a IV de la New York Heart
108Revista Uruguaya de Cardiología Clasificación de las miocardiopatías. Un objetivo, muchas propuestas
Volumen 34 | nº 1 | Marzo 2019 Jorge Estigarribia Passaro
Association (NYHA). Por ejemplo, SA-I en un pacien- 1. El criterio de clasificación
te en riesgo por ser portador genético, sin manifes- Es el punto más crítico y objeto de agudo debate en-
taciones fenotípicas y asintomático, o SC-II en uno tre los defensores de una y otra posición.
con alteraciones estructurales y síntomas en CF II.
La ESC utiliza un criterio primordialmente
Aspectos positivos que los autores señalan acer- morfofuncional con el objetivo expreso de “proveer
ca de este sistema son la flexibilidad y la posibilidad un punto de partida inicial para la investigación
de expansión, de modo de acompañar la evolución clínica”, concediendo importancia secundaria a la
del conocimiento sobre la etiopatogenia de las MP. etiología familiar/genética, que aplica en un paso
Si bien en defensa de esta propuesta se afirma que posterior.
su complejidad no es tan importante como aparenta
Consideramos que el argumento de que la en-
y que su manejo se hace más fluido con la experien-
fermedad es clínicamente relevante solo cuando
cia, implica el uso de una extensa nómina de expre-
se demuestra un fenotipo morfológico es difícil-
siones que resulta difícil de dominar si no se cuenta
mente sostenible. El test genético predictivo “en
con una ayuda escrita o, tal como han elaborado y
cascada” entre los familiares en primer grado de
recomiendan los autores, una aplicación web que
un caso índice determina si se requiere o no una
está disponible en la dirección http://moges.biome-
estrategia de seguimiento, el riesgo de arritmias
ris.com/.
letales en caso de mutaciones “malignas” o de una
Como limitaciones adicionales, se ha señalado elevada carga familiar de muerte súbita y la posi-
que esta clasificación no cumple con los criterios bilidad de transmisión del defecto a la descenden-
diagnósticos de las MP en varias situaciones clíni- cia(28,29).
cas, y no siempre es aplicable en la práctica dada la
La AHA no discrimina patrones anatomofuncio-
carencia de estudios genéticos en muchos centros.
nales; establece una división inicial en MP prima-
Su uso podría resultar más apropiado en ámbitos de
rias y secundarias y clasifica las primarias según su
atención terciaria y particularmente en institucio-
origen genético, adquirido o mixto, para realizar la
nes de diagnóstico e investigación genética, en los
descripción de cada entidad en una etapa posterior
cuales el volumen de pacientes y la formación
en forma individual, asignándolas a cada uno de
específica permitirían obtener la máxima utilidad
esos grupos.
de este sistema.
Una consecuencia del criterio morfológico es
En la tabla 2 se resumen los puntos más destaca-
que reúne dentro del patrón de hipertrofia una mul-
dos en la evolución del conocimiento y de los concep-
tiplicidad de entidades muy heterogéneas, lo que ha
tos que dieron origen a las principales clasificacio-
propiciado un debate en cuanto a nomenclatura y
nes que han sido publicadas.
ubicación taxonómica entre las sociedades europea
y norteamericana(30,31).
La ESC reconoce que históricamente el diagnós-
tico de MCH fue establecido ante una hipertrofia
Comentario
ventricular –en el sentido histopatológico del térmi-
Analizaremos aquí solamente los aspectos confusos no– no explicable por condiciones de carga hemodi-
o controversiales de las clasificaciones de la AHA y námica, generada en mutaciones que afectan a las
la ESC, dado que ambas mantienen vigencia y exhi- proteínas del sarcómero, excluyendo enfermedades
ben diferencias sustanciales tanto en la definición sistémicas como la amiloidosis, patologías por alma-
como en los criterios para el ordenamiento taxo- cenamiento de glucógeno, enfermedades de los liso-
nómico de las MP, en tanto que la clasificación somas, o participación miocárdica en síndromes
MOGE(S) presenta objetivos y metodología especí- multiorgánicos, originadas en mutaciones en otros
ficos diferentes a sus predecesoras. blancos genético-moleculares.
En primera instancia, dos aspectos básicos de No obstante, su posición es la de incluir a todas
coincidencia entre ambos documentos son la incor- estas condiciones bajo el término MCH con un enfo-
poración de los conocimientos adquiridos en el cam- que exclusivamente morfológico y de utilidad prác-
po de la genética molecular, esenciales para su cla- tica, fundamentado en la dificultad en diferenciar-
sificación y para su manejo clínico, y la exclusión las de la verdadera hipertrofia por estudios de ima-
del grupo de las MP de la enfermedad miocárdica gen, el dudoso rendimiento y eventual riesgo de la
originada por HTA, EC, VP y CC, punto tratado en biopsia endomiocárdica y la necesidad de armonizar
forma desconcertante en la clasificación de la criterios con la práctica pediátrica habitual, que in-
WHO/ISFC de 1995. cluye estas enfermedades por acumulación o infil-
Los temas controversiales incluyen: tración bajo la denominación de MCH.
109También puede leer

















































