Control de Calidad de procesos y producto acabado - Vicente Merino Bohórquez UGC Farmacia. HU Virgen Macarena (Sevilla) 18 de Octubre de 2017 - SEFH
←
→
Transcripción del contenido de la página
Si su navegador no muestra la página correctamente, lea el contenido de la página a continuación

Control de Calidad de
procesos y producto
acabado.
Vicente Merino Bohórquez
UGC Farmacia. HU Virgen Macarena (Sevilla)
18 de Octubre de 2017
Índice
1. Control de calidad fisicoquímico:
• Control de contenido en principio activo
• Control de pH
• Control de osmolalidad
• Controles de partículas
• Controles graviméticos
2. Control de calidad microbiológico en productos no estériles:
• Pruebas de promoción del crecimiento y aptitud del método de recuento
• Pruebas de recuento microbiano
• Pruebas de microorganismos específicos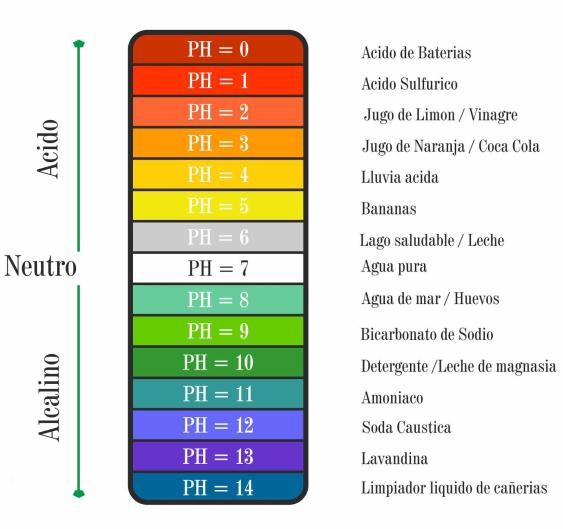
Control de calidad: concepto
• El Control de calidad forma parte de las Normas de Correcta Fabricación de
Medicamentos:
concierne al muestreo, las especificaciones y el control
así como a los procedimientos de organización, de documentación y de liberación que
garantizan que los análisis necesarios y adecuados han sido realmente efectuados
y que las materias primas, los materiales de acondicionamiento y los productos no se
han liberado para su utilización, venta o distribución sin que su calidad haya sido
juzgada como satisfactoria.
OBJETIVO: elaborar medicamentos EFICACES, SEGUROS y de CALIDAD.
Control de contenido en principio activo • Son ensayos analíticos establecidos para garantizar el contenido en principio activo según farmacopea de una preparación. • Diversas técnicas pueden ser utilizadas, siendo la más universal y ampliamente utilizada la: • CROMATOGRAFÍA LÍQUIDA DE ALTA RESOLUCIÓN (HPLC)
Ejemplo: elaboración cápsulas 10 µg de etinilestradiol
Prescripción de etinilestradiol 10 µg cápsulas Endocrionología
Pediátrica
Estrógeno muy potente derivado del estradiol.
Activo por vía oral
Indicado para el tratamiento del retraso puberal (hipogonadismo
femenino)
Dosis de inicio 2,5-5 µg/día con incrementos de hasta 25 µg/día a
los 2,5 años desde el inicio
Cañete-Estrada R, Mata-Rodriguez C, Aguilar-Quintero M. Retraso puberal. Hipogonadismos. Protoc diagn ter pediatr. 2011;1:205-17.
PNT etinilestradiol cápsulas
Calidad por
diseño
(QbD)
Validación PNT
Con 3 Aplicación de Optimización
Elaboración
elaboradores los criterios de la
de 3 lotes
distintos RFE 2.9.40 formulación
Aparatos
Cromatógrafo de líquidos con detector
UV-DAD (Agilent® Infinity 1260)
Balanza Microanalítica (Mettler ®
Toledo)
Fundamentos HPLC
Técnica de separación de moléculas orgánicas en disolución.
Propiedades hidrofóbicas diferentes de moléculas (HPLC-RP)
Cada molécula aparece TIEMPO DE RETENCIÓN
Permite cuantificación: relación Señal-Concentración
La separación se llevó a cabo en COLUMNA porosa de
Octadecilsilano (C18).Materia Prima
Etinilestradiol (DCI):
•Especificaciones USP 34:
•Riqueza 97-102% de C20H24O2 con respecto sustancia seca.
•Fagron Ibérica S.A.U.Certificado GMP/PhEur R=99.9% m/m.
Celulosa microcristalina 101
(PhEur)
Riboflavina (PhEur)
Cápsulas gelatina duras amarillas nº 4 (PhEur)
•Colorantes (cumple Directiva 231/2012/CE, 2009/35/CE. Libre BSE/TSE)Proceso de Elaboración
Proceso de dilución (“prezmecla”): mortero y pistilo.
- Pesar: 0,1 g de EE + 0,1 g Riboflavina + 19.8 g de
CM Mezclar
- Concentración EE: 5 mg/g de mezcla
Elaboración de la mezcla: mortero y pistilo.
•Pesar 0,2 g de premezcla + 6,8 de CM Mezclar
•Concentración EE: 1 mg en 7 g de Mezcla
Elaboración de las cápsulas1: capsulador manual y barredor
•Introducir los 7 g de la mezcla en el capsulador con 100 cáps. nº 4
•Cantidad EE: 10 µg/cápsula (70 mg de contenido por cápsula)
1Procedimiento elaboración cápsulas duras según FN (PN/L/FF/001/00, Formulario Nacional 1ª Edición)Control analítico: puntos críticos
Operación de
mezclado
Premezcla •Control
(5 mg/g) analítico
Mezcla para
•Control
encapsular (1
analítico
mg en 7 g)
Cápsulas de
•Control
etinilestradiol
analítico
10 µgCondiciones cromatográficas y validación
Validación de la técnica:
Flujo=1,0 mL/min Rango de concentracíon: 0.7-6 µg/mL
Linealidad (n=6) (R2>0.9998)
Fase Móvil: 50/50 AGUA:ACN Exactitud: 0.7, 4 y 6 µg/mL (n=5/conc)
Tª =30ºC R 98.5-101.5%
Vol.inyección= 20 µl Precisión: intra e interdía (5 dias consecutivos) CV
|
| ' '
|
|
(%)Aplicación criterios RFE 2.9.40
• Se define como el grado de uniformidad de la cantidad de sustancia
activa en el conjunto de unidades.
Capítulo
2.9.40.
Uniformidad
• Objetivo asegurar la consistencia de las preparaciones unidosis
de las (cápsulas,comprimidos,…) en un intervalo de la cantidad declarada.
preparaciones
unidosis.
• Para demostrarlo, dos métodos: U. Contenido (UC) y V. Masa (VM)
• En nuestro caso, estamos obligados a realizar UC (Dosis < 25mg)Uniformidad de contenido
• FF sólidas: valorar individualmente 10 unidades Valor aceptación
Χmedia de los contenidos individuales en % de la cantidad declarada (T)
k cte. de aceptabilidad:
- Si nº unidades es 10 2,4
- Si nº unidades es 30 3,0
s desviación típica de la muestra
M Valor de referencia cuya aplicación depende de T (cantidad declarada en la etiqueta en el momento de fabricación):
Caso 1 T ≤101.5 %
Caso 2 T >101.5%
CUMPLE SI VA< 15 y el contenido medio EE 90-115%Cálculo Valor de Aceptación
VALOR DE REFERENCIA (M)
M=X
Si 98,5%≤X≤101,5%
(VA=ks)
M (caso 1)
A aplicar cuando Si X101.5%
(VA=X-101.5+ks)Variación en Masa
• Procedimiento:
a) Pesar individualmente 10 cápsulas de forma exacta (preservando identidad)
b) Vaciar el contenido de cada cápsula
c) Pesar individualmente las cubiertas vacías de forma exacta
d) Calcular la masa neta por diferencia de masa (llena-vacia)
e) Calcular el valor de aceptación idem UC.
f) Calcular el contenido de sustancia activa de cada cápsula a partir de la masa de producto
retirado de las cápsulas individuales y del resultado de la valoración.
Xi: contenido individual de cada cápsula %
wI: masa individual de las unidades analizadas
A: contenido en sustancia activa (%) de cada cápsula analizada
W: masa media de las cápsulas individualesControl
Caps llena (mg)
del Proceso
Casp. Vacía
Contenido (mg)
Unif. Contenido
% Contenido VARIACIÓN MASA
(mg)
(analizado)
• Formulación
105.1 original:
41 64.1 77.24 71.85
• Muestreo41.3
111.8 de 5 zonas 70.5 86.37 88.37
111.3 40.6 70.7 85.47 87.69
111.4 Análisis 40.3 Premezcla
71.1(% R) Mezcla (% R)
86.50 89.25
109 ZONA 1 40 69
93.01 83.55 88.31 83.65
109.7 40.1 69.6 81.17 89.43 81.98
ZONA 2 93.04 2
107.2 40.6 1 66.6 81.36 78.63
106 ZONA 3 38.9 93.04
5
67.1 88.21
78.59 76.53
111.7 ZONA 4 40.1 92.71
71.6 4 87.92 88.62 91.35
109.1 40.3
3 68.8
ZONA 5 94.87 84.45 87.51 84.31
MEDIA 83.26 83.36
MEDIA 93.33 88.41
90-115% NO CUMPLE S 3.56 6.20
• Contenido
VA>15 Medio: 90-115%
NO CUMPLE EE
Valor Aceptación 23.78 30.02
Solución: pesar un exceso del 10% en EE.Optimización de la fórmula
Optimización de la Fórmula
• Caps
Formulación
llena (mg)
modificada
Casp. Vacía (10% exceso EE):
Contenido (mg)
% Contenido
VARIACIÓN EN MASA
(mg) (analizado)
• Muestreo de 5 zonas
109 40.9 68.1 95.70 92.24
114 Análisis
39.8 Premezcla (%
74.2 R)
103.48 Mezcla 108.68
(% R)
111.4 41.7 69.7 98.14 96.82
ZONA 1 98.21 96.98
111.6 39.8 71.8 102.32 103.98
112.3 ZONA
40.6 2 71.7 98.08 101.68 97.85
103.19
1 2
110.5 40.5 70 98.98 98.06
ZONA 3 98.98
5
98.27
108.4 40.2 68.2 102.06 98.52
108.5 ZONA
40.5 4 68 98.94 99.17
4 98.4695.45
114 40.8
3 73.2 102.32 106.01
ZONA 5 97.30 98.75
112.2 40.6 71.6 101.16 102.52
MEDIA
MEDIA 98.31 100.50 98.06
100.55
90-115% CUMPLE S 2.41 5.13
• Cumple contenido 90-115%
L1Conclusiones
Se ha demostrado que en dosificaciones pequeñas añadir
un exceso nos ayuda a cumplir con los atributos de calidad
exigidos.
En elaboración es necesario identificar puntos críticos
para un buen control del proceso.
Son necesarios más estudios que validen los
Procedimientos Normalizados Trabajo de formulaciones
que tengan un mayor impacto asistencialEVALUACIÓN DE LA PREPARACIÓN DE
ANTIBIÓTICOS INTRAVÍTREOS EN
URGENCIAS OFTALMOLÓGICAS
Autores: Merino-Bohórquez V1, Vázquez-Alfageme C2,Fernández-Pérez B2, Cordero-Ramos J1,
Donoso-Rengifo C1, Ponte-Zuñiga B2, Cameán M1.
1UGC Farmacia. H.U. Virgen Macarena. Sevilla.
2UGC Oftalomología. H.U. Virgen Macarena. Sevilla.Introducción
Endoftalmitis infecciosa reacción inflamatoria intraocular
• Colonización de bacterias, hongos o raramente parásitos.
• Es una complicación grave perdida visión y compromete intregridad globo ocular
Causa principal de endoftalmitis infecciosa exógena cirugía ocular
90 % cirugía de cataratas
Es clave realizar diagnóstico clínico y microbiológico precoz.
Objetivo obtener concentraciones ATB en vítreo rápidamente desde el diagnóstico.
Destacar la tendencia al tratamiento quirúrgico (vitrectomia)Objetivo y diseño
Hipótesis:
La preparación de los antibióticos intravítreos en consulta en condiciones de
urgencia, no es idónea, tanto por la dificultad de mantener condiciones estériles en
un medio no preparado para ello, como muchas veces por falta de experiencia en el
manejo de inyecciones intravítreas del personal.
Objetivo principal:
Determinar si la concentración de los antibióticos (ceftazidima y vancomicina) para
administración intravitrea, preparados en la consulta de urgencias por oftalmólogos,
es la que se especifica en la USP 34.
Diseño:
- Estudio prospectivo, descriptivo y observacional
- Valorar el éxito de las preparaciones de antibióticos realizadas por una muestra de
especialistas en oftalmología durante un periodo de 4 meses.Protocolo de preparación AHVM1,2
Ceftazidima 20 mg/mL Vancomicina 10 mg/mL
Disolver el contenido de un vial de 1 g con Disolver el contenido de un vial de 500 mg
5 mL de suero fisiológico, extraídos de una con 5 mL de suero fisiológico, extraídos de
bolsa de 50 mL NaCl 0,9%. una bolsa de 50 mL NaCl 0,9%.
Devolver el contenido del vial totalmente Devolver el contenido del vial totalmente
disuelto a la bolsa de suero para obtener disuelto a la bolsa de suero para obtener una
una concentración final de 20 mg/mL. concentración final de 10 mg/mL.
Extracción de 2 mL de ambas soluciones para
cuantificación por HPLC
1Guía de práctica clínica de la ECRS (Sociedad Europea de Cirujanos de Catarata y Refractiva). www.escrs.org
2Guía de práctica clínica de la SERV (Sociedad Española de Retina y Vítreo) www.serv.esCromatografía y validación: Ceftazidima
mAU
1.691
120
100
Validación de la técnica:
80 Rango de concentracíon: 0,1-0,3 mg/mL
Flujo=1,3 mL/min (n=6)
Linealidad (R2>0.9996)
60 Fase Móvil: 80/20
Exactitud: 0.1, 0.2 y 0.3 mg/mL (n=5)
buffer (pH=3.5, 0.01M):MeOH
R 98.5%-102%
40 Tª =30ºC
Precisión: intra e interdía (5 dias
Vol.inyección= 5 µl consecutivos) CV (%)Cromatografía y validación: Vancomicina
Validación de la técnica:
Flujo=1,3 mL/min Rango de concentracíon: 0,05-0,15 mg/mL (n=6)
Linealidad (R2>0.9998)
Fase Móvil: 80/20
Exactitud: 0.05, 0.1 y 0.15 mg/mL (n=5)
buffer (pH=3.5, 0.01M):MeOH R 98.5%-102%
Tª =30ºC Precisión: intra e interdía (5 dias consecutivos)
CV (%)Resultados (I)
Participaron 24 oftalmólogos 129 preparaciones:
64 IVT de Ceftazidima 20 65 IVT de Vancomicina 10
mg/mL mg/mL
Datos demográficos:
Experiencia media 90 % se definió 27% se definió
Media de edad: especialidad: como “habituado a como “habituado al
45,40 ± 13,47 años.
17,88 ± 12,75 años la cirugía” manejo IVT”Resultados (II)
Nºpreparaciones
preparaciones
CONTROLES
Nº preparaciones
Nº por
DE
por concentración
COMPARACIÓN
VANCOMICINA 10 mg/mL
concentración
Nº preparaciones CEFTAZIDIMA
CEFTAZIDIMA
VANCOMICINA
% RECUPERACIÓN (N=3)
16
12
14 CEFTAZIDIMA VANCOMICINA
10 PROFESIONAL
preparadas
12 27% (media ± DE) (media± DE)
preparadas
108
45% DUE 1 36%
8 97.51 ± 1.8150%
% 98.12
120% Conc.
Conc.
FARMACEÚTICO 1 98.20 ± 1.20 % 99.2 ± 1.43Nominal
Nominal
%
44
23% 19% 90-120% Conc. Nominal
2
DUE 2 99.1 ± 1.23 % 102 ± 1.02 %
02
0 FARMACÉUTICO 2 101,1 ± 1.78 98 ± 1.15 %
>30 40 50 60 70 80 90 100 110 120 130 140 o
mayor
Intervalos
Intervalosde
deConcentración
Concentración(%)
(%)Conclusiones
La preparación de IVT en servicios de
Urgencias de Oftalmología no cumple
con las especificaciones de
concentración de la USP.
Los Servicios de Farmacia deben de
hacerse cargo de la elaboración de las
IVT no sólo por cuestiones de
esterilidad sino porque el personal de
farmacia tiene más experiencia en el
manejo de este tipo de preparaciones.Control de pH
Parámetro
crítico que
mide la
actividad del
ion hidrógeno
Alto impacto Es de gran
en la calidad y importancia
estabilidad de tenerlo
la presente en la
formulación. elaboración.Objetivo Tener en
•Excipientes
(conservantes)
cuenta pH influyen en pH
vehículos •Caracterizar pH de los
vehículos a utilizar
pH
estabilidad
principios
activos:
Elaboración de FM
oral ¿pH máxima
estabilidad?
Merino-Bohórquez V, García Palomo M, Portillo Vázquez S, Murillo Izquierdo M, Ávila Álvarez JR, Cameán Fernández M. CONTROL DEL pH EN LA ELABORACIÓN DE VEHÍCULOS
LÍQUIDOS ORALES: ¿REALMENTE SON IMPORTANTES LOS EXCIPIENTES?. Comunicación ORAL. 60 Congreso SEFH.pH de estabilidad de principios activos
Principio activo Intervalo pH estabilidad
Midazolam 2.5-3.7
Propranolol 2.8-4.0
Hidralazina 3.0-5.0
Flecainida 3.8-4.8
Clonidina 3.5-5.5
Ranitidina 6.7-7.5
Furosemida 7.0-10.0
Fenobarbital 9.2-10.2
The United States Pharmacopeia, 34nd rev., and The national formulary, 29th ed.Rockville, MD: United States Pharmacopeial Convention; 2011.Material y métodos:
• Se elaboraron cuatro lotes de cuatro tipos vehículos líquidos orales:
Jarabe simple (64 partes sacarosa/36 partes de agua)
Jarabe simple con conservantes parahidroxibenzoato de
metilo y propilo (Nipagin® BASE al 0.06% p/v y Nipasol®
BASE 0.04%)
Jarabe simple con conservantes parahidroxibenzoato de
metilo sódico y propilo sódico (Nipagín® SÓDICO 0.06%
p/v y Nipasol® SÓDICO 0.04% p/v)
Solución de sorbitol al 70% con conservante (sorbato
potásico 0.15 % p/v) csp. 100 mLElaboración
Cada vehículo se
elaboró:
pHmetro
Dos tipos de Dos tipos de Medida del pH
CRISON® GLP21+
agua: azúcar (jarabes): por triplicado
(Tª 23.5ºC)
Ultrapurificada Azucar
(tipo Milli-Q) doméstico
Purificada Sacarosa PhEur
(Grifols®) (Fagron®)SOLUCIÓN
A. Jarabe simple
DEB.MIDAZOLAM
Jarabe simple con
1 mg/mL
C. Jarabe simple con
Componentes/ D. Solución de sorbitol
Resultados
conservantes conservantes
(64 partes
pH media±DE parahidroxibenzoato de parahidroxibenzoato de al 70% con sorbato
sacarosa/36 partes metilo y propilo base metilo sódico y propilo sódico
potásico 0.15 % p/v
de agua) (0.06% p/v-0.04%) (0.06% p/v-0.04% p/v)
AGUA PURIFICADA PhEur +
AZUCAR DOMÉSTICO
6.89±0.050 6.32±0.017 8.79±0.005
6.44±0.005
AGUA PURIFICADA PhEur + (EN AGUA PURIFICADA PhEur)
SACAROSA PhEur
6.60±0.005 5.69±0.015 8.71±0.010
AGUA ULTRAPUFICADA
6.87±0.08 6.41±0.010 8.75±0.010 6.45±0.005
MILLI-Q + SACAROSA PhEur
(EN AGUA ULTRAPURIFICADA
A
AGUA ULTRAPUFICADA B C
MILLI-Q + AZUCAR 6.49±0.051 6.06±0.026 8.90±0.050 MILLI-Q)
DOMÉSTICO
pH AGUA ULTRAPURIFICADA
MILLI-Q
6.58±0.010
pH AGUA PURIFICADA PhEur 5.56±0.010Jarabe simple COMERCIAL
Fabricante Guinama® Acofarma® Fagron®
Componentes % Composición % Composición % Composición
Sacarosa 64 64 64
Agua purificada 36 36 36
Metil paraben 0,06 (sódico) - 0,1 (base)
Propil paraben 0,04 (sódico) - -
Sorbato potasico - c.s. -
Benzoato sódico
Ácido cítrico
pH 7-9 5.5 4.4-4.6Conclusiones
Para principios Para principios activos
cuya estabilidad máxima
Vehículos elaborados con: El tipo de agua
activos sean sea en medio ácido
•Azúcar doméstico en general,
tiene pH más alcalino frente a los no influye en la
estables a pH podrían utilizarse A, B y D vehículos de sacarosa PhEur
básico se debería según convenga y un probablemente por las impurezas elaboración de
que aporta.
utilizar el vehículo ácido débil (p.e., ácido •Agua Milli-Q tienen un pH más sorbitol como
cítrico) para alcanzar el alcalino que los de agua
C. pH óptimo. purificada probablemente por vehículo.
tener un pH más cercano a la
neutralidad que el agua
purificada.Ejemplos: Fenobarbital solución 10 mg/mL
% CONTENIDO EN PRINCIPIO ACTIVO FENOBARBITAL
FORMULAICONES CON CONSERVANTES
Evolución pH fenobarbital (CON CONSERVANTES)
10
F1 40ºC F1 25ºC F1 5ºC
110 9,5
100
90
9
80
% RECUPERACIÓN
70
pH
8,5
60
50
8
40
30
20
7,5
10
0 7
DIA 0 DIA 5 DIA 7 DIA 14 DIA 21 DIA 28 DIA 42 DIA 50 DIA 60 DIA 90 DIA 0 DIA 5 DIA 7 DIA 14 DIA 21 DIA 28 DIA 42 DIA 50 DIA 60 DIA 90
- pH estabilidad 9.2-10.2
- pHControl de osmolalidad
• Es una propiedad coligativa que depende del número de moléculas en una disolución
relacionada con la presión osmótica.
• La osmolalidad de una solución es una cantidad teórica expresada en osmoles por Kg
(Osmol por Kg) de una solución.
• La osmolalidad de la sangre oscila 285-310 mOsm/Kg
• El osmómetro permite calcularla a través del descenso del punto de congelación de una
solución.
• Su control resulta útil en:
• Soluciones orales (pediátricas-neonatales)
• Colirios
• Preparados intravenosos (riesgo de flebitis química)Ejemplos de osmolalidad de soluciones
Clonidina 20 mcg/mL sol. oral
1350 mOms/Kg
Hidralazina 10 mg/mL sol. oral
1398 mOsm/Kg
Fenobarbital sódico 10 mg/mL
sol. oral 2916 mOms/Kg
Isoniazida 50 mg/mL sol. oral
2890 mOsm/Kg
Etambutol 50 mg/mL sol. oral
2375 mOsm/Kg
Solución Nefroprotectora
Solución intravenosa
582 mOsm/KgControl de partículas (I)
a) Visibles:
• Objetivo: detección de partículas extrañas, móviles no disueltas,
distintas de las burbujas de un gas y que están presente
involuntariamente en las soluciones.
• Aparato: cubículo de luz con intensidad adecuada con fondo
negro y blanco sobre el que se invierte la solución para
inspeccionar partículas visibles.Control de partículas (II)
b) Sub-visibles:
• Contaje de partículas subvisibles en soluciones inyectables para
uso parenteral (reconstituidas o no) exentas de partículas.
• Métodos:
1. Método de obscuración de la luz:
• Partículas sólidas y líquidas en suspensión.
• Si no cumple los límites o por razones técnicas (viscosidad) se pasa a
método 2.
• Grandes volúmenes.
2. Método de Microscopía óptica: microscopio binocular,
iluminadores y retícula de medición de diámetro de partículas.Controles gravimétricos
• ENSAYO DE UNIFORMIDAD DE MASA (RFE 2.9.5)
- Objetivo: para garantizar la uniformidad de las unidades de dosificación,
cada unidad en un lote debe tener un contenido de fármaco dentro de un
intervalo estrecho alrededor de la cantidad declarada.
- Método:
1. Pesar individualmente 20 unidades tomadas al azar o, para las preparaciones unidosis
presentadas en envases individuales, el contenido de 20 unidades, y determinar la masa
media.
2. No más de 2 de las 20 masas individuales se desvían de la masa media en un porcentaje
más elevado que el porcentaje de desviación indicado en la Tabla 2.9.5.-1. y ninguna se
desvía en más del doble de ese porcentaje.Tabla 2.9.5-1
Forma farmacéutica Masa media Porcentaje de desviación
Comprimidos (no recubiertos y 80 mg o menos 10
recubiertos con película)
Más de 80 mg y menos de 250 mg 7,5
250 mg o más 5
Cápsulas, granulados (no Menos de 300 mg 10
recubiertos, unidosis) y polvos
300 mg o más 7,5
(unidosis)
Polvos para administración Más de 40 mg 10
parenteral* (unidosis)
Supositorios y óvulos Todas las masas 5
Polvos para colirios y polvos para Menos de 300 mg 10
baños oculares (unidosis) 300 mg o más 7,5
* Cuando la masa media es igual o inferior a 40 mg, la preparación no se somete al ensayo de uniformidad de
masa, sino al ensayo de uniformidad de contenido de las preparaciones unidosis (2.9.6).Ejemplo práctico: cápsulas dexametasona 10 mg
Ejemplo práctico: cápsulas dexametasona 40 mg
Control microbiológico no estériles (USP y ) (I)
• Prueba de promoción del crecimiento y aptitud del
método de recuento:
Demostrar la capacidad de crecimiento de cepas
patrón en la formulación del estudio.
S. aureus ATCC 6538, P. aeruginosa ATCC 9027, B. subtilis
ATCC 6633, C. albicans ATCC 10231, A. brasiliensis ATCC
16404, E. coli ATCC 8739, S. enterica ATCC 14028 y C.
sporogenes ATCC 11437.Control microbiológico no estériles (USP y ) (II)
• Pruebas de recuento microbiano en placa
Recuento total de microorganismos aerobios (RTMA) y el recuento total
combinado de hongos filamentosos y levaduras (RTCHL) que podían
desarrollarse en condiciones aerobias.
Límite: RTMA de 102 y RTCHL 101 y debe haber ausencia de E. coli.Control microbiológico no estériles (USP y ) (III)
• Pruebas de crecimiento de microorganísmos específicos
Demostrar la ausencia o presencia limitada de microrganismos
específicos.
Detección de bacterias gram negativas tolerantes a bilis: E. coli,
Salmonella, P. aeruginosa, S.aureus, Clostridios, C. albicans.Estudio de estabilidad fisicoquímico y microbiológico
de clonidina en solución.
Objetivos
I. Estudio de la estabilidad fisicoquímica de clonidina solución
En diferentes condiciones de conservación durante un período de tiempo
de hasta 90 días siguiendo las recomendaciones de las guías ICH.
Las condiciones a estudiar serán tres:
Refrigeración (2-8ºC)
Temperatura ambiente (25±2ºC)
40 ºC (±2ºC)
II. Estudio microbiológico según USP
Envases cerrados
Envases abiertosMetodología: Elaboración Elaboración de las fórmulas: • Fórmula 1(CON conservante): • Clonidina clorhidrato SM (1 mg/mL)………………...2 mL • Agua ultrapura………………………………….…..….…48 mL • Sorbato potásico……………………………………….….150 mg • Jarabe simple (sacarosa/agua UP 64:36) csp…100 mL • Ajustado a pH=4-5 con ácido cítrico 25%. • Fórmula 2 (SIN conservante): • Clonidina clorhidrato SM (1 mg/mL)…………….…..….2 mL • Agua ultrapura……………………………………………..…48 mL • Jarabe simple (sacarosa/agua UP 64:36) csp……..100 mL • Ajustado a pH=4-5 con ácido cítrico 4%.
Metodología: condiciones almacenamiento
Elaboración de 18 botes de vidrio topacio:
Solución madre
F1: Fórmula CON conservantes F2:Fórmula SIN conservantes
1 mg/mL
LOTE A: 3 BOTES A 40ºC LOTE A: 3 BOTES A 40ºC 3 BOTES A 5ºC
LOTE B: 3 BOTES A 25ºC LOTE B: 3 BOTES A 25ºC
LOTE C: 3 BOTES A 5ºC LOTE C: 3 BOTES A 5ºCResultados microbiológicos: envases NO esterilizados
CERRADOS Día 0 Día 7 Día 14 Día 28 Día 42 Día 60 Día 90
F1 40ºC
Bacterias y hongos - -
F1 25ºC Bacterias - - - - - -
Bacterias - -
F1 5ºC
F2 40ºC Hongos - - -
F2 25ºC
Bacterias -
Bacterias - - - - - -
F2 5ºC Bacterias -
Bacterias -Resultados microbiológicos: ESTERILIZADOS
Resultados microbiológicos: envases esterilizados
CERRADOS Día 0 Día 7 Día 14 Día 28 Día 42 Día 60 Día 90
F1 40ºC
F1 25ºC
F1 5ºC
F2 40ºC
F2 25ºC
F2 5ºC
“Somos lo que hacemos
día a día. De modo que la
excelencia no es un acto,
sino un hábito”.
AristótelesTambién puede leer

















































