INFECCIONES VIRALES E INMUNODEFICIENCIAS PRIMARIAS (IDPs)
←
→
Transcripción del contenido de la página
Si su navegador no muestra la página correctamente, lea el contenido de la página a continuación
XI Curso de
Infectologia
Pediátrica
INFECCIONES VIRALES E
INMUNODEFICIENCIAS
PRIMARIAS (IDPs)
ANDREA MARTÍN NALDA, PERE SOLER-PALACÍN
Unitat de Patologia Infecciosa i Immunodeficiències de Pediatria
Hosp. Universitari Vall d´Hebron. Barcelona
Barcelona 9 de febrero de 2012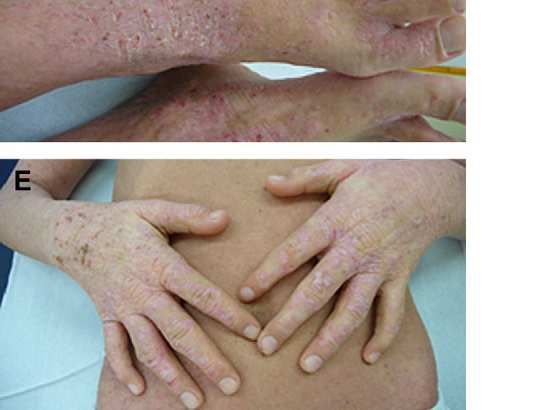
¿QUÉ SON LAS IDP?
Patogenia Mutaciones en genes que codifican ciertos
componentes del Sistema Inmune
A menudo hereditarias (AR/X linked)
Actualmente > 200 defectos genéticos asociados a IDP
1 : 2000 RN vivos
Defectos humorales > 50% de las IDP
Clínica: INFECCIONES (bacterianas, virales, hongos…)
Autoinmunidad
Neoplasias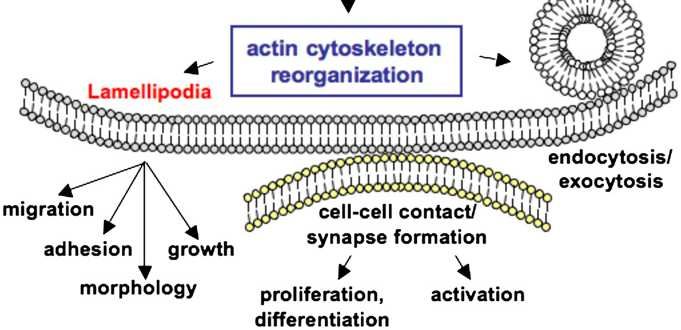
Factores a tener en cuenta a la hora de orientar al
paciente con sospecha de IDP
HISTORIA FAMILIAR
EDAD COMIENZO INFECCIÓN
TIPO DE MICROORGANISMO
EXPLORACIÓN FÍSICA
MANIFESTACIONES ÓRGANO-ESPECÍFICAS
PRUEBAS DE LABORATORIO
TIPO DE MICROORGANISMO
Serratia marcescens, Salmonella spp, Enfermedad
Staphylococcus aureus, Aspergillus spp granulomatosa crónica
Sepsis por Streptococcus pneumoniae Déficit de NEMO
Déficit de IRAK4
S. aureus Hiper IgE
VEB Sd linfoproliferativo ligado a X (XLP)
Virus papiloma humano Sd WHIM
Infecciones graves por virus IDP combinadas
respiratorios e intestinales
Giardia lamblia IDP de tipo humoral
Enterovirus ALX
Pneumocystis jiroveci IDP combinadas
Déficit de NEMO
VHS Déficit TLR3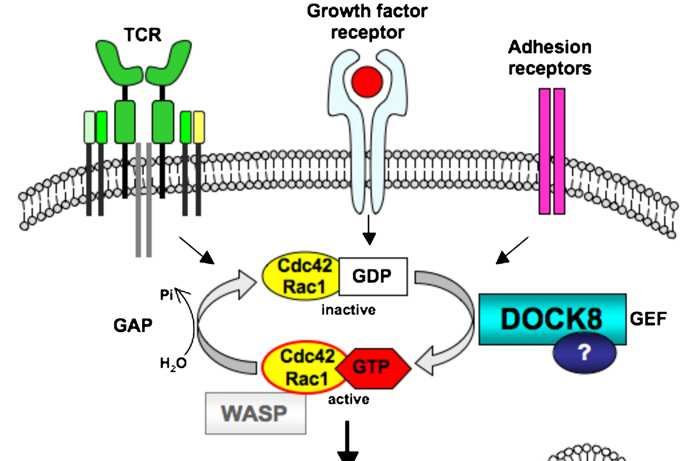
CASO CLINICO 1

CASO 1
Niño de 4 meses que ingresa en UCI-P por
insuficiencia respiratoria
• AF: Consanguinidad. Padres de origen Marroquí. Antecedente
de un hermano fallecido (causa desconocida) en los primeros
meses de vida.
• AP:
-RN a termino. Peso adecuado al nacimiento
-Diarrea intermitente desde el primer mes de vida. Retraso
ponderal.
-Unos días antes del ingreso comienza con tos, febrícula y
disminución de la ingesta. Empeoramiento progresivo.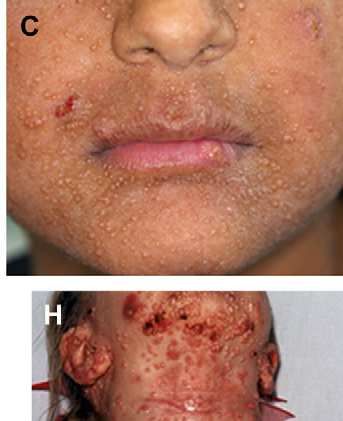
CASO 1
Neumonía intersticial
BAL: virus
Parainfluenza - 3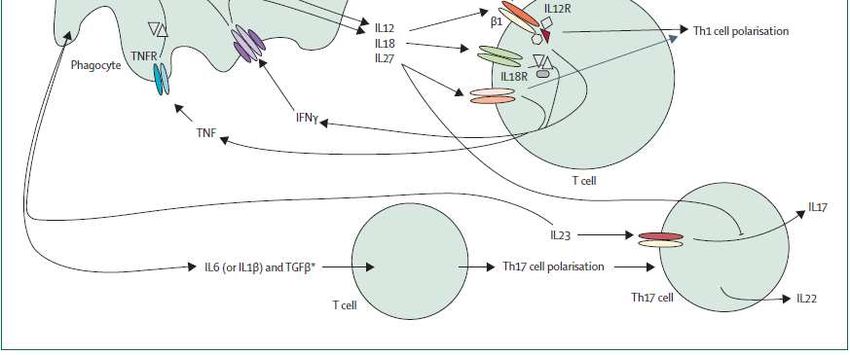
¿Consideran adecuado llevar
a cabo un estudio inmunitario
en este momento?
¿Consideran adecuado llevar a cabo un estudio inmunitario en este momento? 1. No, no cumple criterios para indicarlo. 2. Si, ha presentado una infección grave + retraso ponderal 3. Si ya que siempre una infección pulmonar tiene que estudiarse inmunológicamente 4. No, porque ha tenido mala suerte y cualquier virus puede producir en el lactante una infección grave
¿Consideran adecuado llevar a cabo un estudio inmunitario en este momento? 1. No, no cumple criterios para indicarlo. 2. Si, ha presentado una infección grave + retraso ponderal 3. Si ya que siempre una infección pulmonar tiene que estudiarse inmunológicamente 4. No porque ha tenido mala suerte y cualquier virus puede producir en el lactante una infección grave
SOSPECHA DE IDP: Signos de alarma (1)
1 > 8 OMA en un año
2 > 2 neumonías (confirmadas radiológicamente) en un año
3 > 2 sinusitis en un año
4 > 2 infecciones tejidos profundos en un año o de localización no
habitual
Infecciones recurrentes cutáneas profundas o abscesos viscerales
5
6 Necesidad frecuente de usar antibioterapia endovenosa para curar
infecciones
7 Infecciones por organismos no habituales u oportunistas
8 Historia familiar de inmunodeficiencias o infecciones recurrentesSOSPECHA DE IDP: Signos de alarma (2)
9 Retraso ponderoestatural
10 2 o más meningitis o infecciones graves
11 Fenómenos autoinmunes frecuentes
12 Muguet o candidiasis cutánea en paciente mayor de un año
13 Rasgos dismórficos asociados a infecciones frecuentes
14 Infecciones postvacunales en vacunas a virus vivos
15 Retraso en la caída del cordón umbilical (> 4 semanas de vida)
16 Ig E > 2000 UI/L sin otra causa aparente (sobre todo con afectación cutánea
e infecciones de repetición)
17 Aftas orales recurrentes
18 Fiebre con sospecha de periodicidad
19 Bronquiectasias sin causa aparente¿Qué pruebas solicitarían?
¿Qué pruebas solicitarían? 1. Hemograma y bioquimica 2. Hemograma e inmunoglobulinas 3. Hemograma, inmunoglobulinas e inmunofenotipo y serología a VIH. 4. Hemograma + test de oxidación
¿Qué pruebas solicitarían? 1. Hemograma y bioquímica 2. Hemograma e inmunoglobulinas 3. Hemograma, inmunoglobulinas e inmunofenotipo y serología a VIH. 4. Hemograma + test de oxidación
CASO 1
Niño de 4 meses que ingresa en UCI-P por
insuficiencia respiratoria
• EC:
- hemograma: Hb 10,2 g/dl, 2900 leucocitos
totales/mm3 (1500 N, 900 L), 198.000 plaquetas/mm3
- Ig G 120mg/dL Ig A 15 mg/dL Ig M 20 mg/dL
- Inmunofenotipo:¿Hay algún valor alterado?
¿Hay algún valor alterado? 1. Ninguno 2. Disminución de IgG 3. Linfopenia, disminución de IgG y de linfocitos T CD3+ y CD19+ 4. Solo linfopenia y disminución de IgG
¿Hay algún valor alterado? 1. Ninguno 2. Disminución de IgG 3. Linfopenia, disminución de IgG y de linfocitos T CD3+ y CD19+ 4. Solo linfopenia y disminución de IgG
VALORES DE REFERENCIA
Ig G (mg/dL) Ig A (mg/dL) Ig M (mg/dL)
RN (término) 610 - 1.540 1-4 6 - 30
3 MESES 170 - 560 5 - 50 30 - 100
6 MESES 200 - 670 8 - 70 30 - 100
1 AÑO 330 - 1.160 10 - 100 40 - 170
2-6 AÑOS 400 - 1.100 10 - 160 50 - 180
7-12 AÑOS 600 - 1.230 30 - 200 50 - 200
ADULTOS 700 - 1600 70 - 400 40 - 230VALORES DE REFERENCIA
RN 1-2m 2-5m 5-9m 9-15m 15-24m 2-5a 5-10a 10-16 Adultos
término
4.100 6.700 5.900 6000 5.500 5600 3.300 2.800 2.200 1.800
LINFOCITOS
(2.200-6.900) (4000-11.300) (3.900-8.600) (4000-9000) (3.100-8.700) (3.400-8.900) (2.300— (1.600-4.300) (1.300-3000) (1.100-2.500)
TOTALES 5.600)
2.500 4.600 3.600 3.800 3.400 3.500 2.300 1.900 1.500 1.200
LINFOCITOS
(1.100-4.200) (2.800-6.500) (2.400-5.600) (2.700-6.100) (1.800-5.900) (2.200-5.500) (1.400-3.600) (1.100-2.800) (1.000-2.000) (700-1.900)
T (CD3+)
1.800 3.500 2.500 2.800 2.300 2.200 1.300 1000 800 700
LINFOCITOS
(800-2.900) (2.100-4.900) (1.600-4.200) (1.700-4.100) (1.300-4.100) (1.100-3.600) (700-2000) (500-1.800) (500-1.300) (400-1.300)
T(CD3+/CD4+)
800 1000 1000 1.100 1.100 1.200 800 800 400 400
LINFOCITOS
(300-1800) (500-1.600) (700-1.500) (700-1.800) (500-1.600) (500-1.800) (500-1.400) (400-1.200) (300-800) (200-700)
T(CD3+/CD8+)
500 1000 1.300 1.300 1.400 1.300 800 500 300 200
LINFOCITOS
(200-1000) (300-1700) (800-2.600) (800-2.200) (700-2.400) (900-2.500) (400-1.500) (300-700) (200-500) (100-400)
B(CD19+)
CÉLULAS NK 800 500 300 300 400 400 400 300 300 300
(200-1800) (300-800) (200-900) (200-800) (200-900) (100-1.100) (100-700) (100-600) (100-700) (100-400)
(CD3+/CD56+CD16
CON LA AUTORIZACIÓN DE J.J.M.van.Dongen (Departments of immunology,Pediatrics,and Epidemiology and Biostatistics Erasmus MC,University Medical Center)¿Es sugestivo de alguna IDP?
¿Es sugestivo de alguna IDP? 1. No, son hallazgos que pueden estar alterados en el contexto de la infección 2. Inmunodeficiencia humoral 3. Defecto fagocitosis 4. Inmunodeficiencia combinada grave (IDCG)
¿Es sugestivo de alguna IDP? 1. No, son hallazgos que pueden estar alterados en el contexto de la infección 2. Inmunodeficiencia humoral 3. Defecto fagocitosis 4. Inmunodeficiencia combinada grave (IDCG)
IDCG
CLASIFICACIÓN
T- B- NK - Dèficit de ADA.
Disgenèsia reticular
T- B - NK + Dèficit de RAG 1, RAG 2
Síndrome de Omenn.
SCID Navajo.
T+ B + Dèficit de IL-2.
Dèficit de ZAP-70
T - B + NK - SCID ligada a X
Dèficit de JAK3.
Dèficit de PNP.
T - B+ NK + Dèficit de IL-7R alfa.
Dèficit de CD3 gamma.Para el diagnóstico definitivo de IDCG en este paciente, ¿qué prueba faltaría?
Para el diagnóstico definitivo de IDCG en este paciente, ¿qué prueba faltaría? 1. Un test de oxidación alterado 2. Una función linfoproliferativa T disminuida 3. Una mala respuesta a vacunas 4. Una alteración en los receptores TLR
Para el diagnóstico definitivo de IDCG en este paciente, ¿qué prueba faltaría? 1. Un test de oxidación alterado 2. Una función linfoproliferativa T disminuida 3. Una mala respuesta a vacunas 4. Una alteración en los receptores TLR
IDCG
DIAGNÓSTICO DE
LABORATORIO
Linfopenia
Linfopenia T (CD4, CD8) ES UNA
Disminución función
linfoproliferativa (PHA)
EMERGENCIA
Disminución Igs (atención a PEDIATRICA!!
‘cifras límite’, IgG maternas).Nuestro paciente
¿Cuál es el tratamiento curativo de una IDCG?
¿Cuál es el tratamiento curativo de
una IDCG?
1. Tratamiento periódico con GG + profilaxis AB
2. No existe
3. TPH o terapia génica o tratamiento sustitutivo
enzimático
4. Solo profilaxis antibiótica, antifúngica y antiviral¿Cuál es el tratamiento curativo de
una IDCG?
1. Tratamiento periódico con GG + profilaxis AB
2. No existe
3. TPH o terapia génica o tratamiento sustitutivo
enzimático
4. Solo profilaxis antibiótica, antifúngica y antiviralCASO CLINICO 2
CASO 2
Niño de 12 años que ingresa en UCI-P por hepatitis
fulminante
• AF: tío (hermano de su madre fallecido a los 20 años por linfoma)
• AP: sin interés. Niño sano hasta este ingreso
• Historia actual: cuadro catarral los días previos y leve
odinofagia. Febrícula acompañante. Ictericia 24 horas antes del
ingreso
Analítica al ingreso:
- AST/ALT: 3000/4100. Coagulopatía. Hipoalbuminemia. Acidosis
- LinfocitosisCASO 2
Niño de 12 años que ingresa en UCI-P por hepatitis
fulminante
• Se descartan medicamentos y otros tóxicos
• Se descarta metabolopatía
• Estudio virus hepatitis : negativo
• Estudio VEB: serologías compatibles con infección aguda y
PCR a VEB positiva en sangre
MONONUCLEOSIS FULMINANTE¿Consideran adecuado llevar
a cabo un estudio inmunitario
en este momento?¿Consideran adecuado llevar a cabo un estudio inmunitario en este momento? 1. No, no cumple criterios para indicarlo. 2. Si, ha presentado una infección grave por un virus que habitualmente no produce esta clínica 3. No, porque hasta ahora era un paciente sano y no hay antecedente de infecciones previas 4. Si, porque todas las hepatitis hay que estudiarlas inmunológicamente
¿Consideran adecuado llevar a cabo un estudio inmunitario en este momento? 1. No, no cumple criterios para indicarlo. 2. Si, ha presentado una infección grave por un virus que habitualmente no produce esta clínica 3. No , porque hasta ahora era un paciente sano y no hay antecedente de infecciones previas 4. Si, porque todas las hepatitis hay que estudiarlas inmunológicamente
¿ PENSARIAN EN ALGUNA
ENFERMEDAD LIGADA A X POR LOS
ANTECEDENTES FAMILIARES?
SÍ
XLPXLP (X-linked lymphoproliferative disease) • Incidencia: 1-3 casos /1000.000 varones. Descrita en 1969. • Manifestaciones clínicas:
XLP (X-linked lymphoproliferative disease)
• Patogenia: Inapropiada respuesta Th2 a infección por VEB y
aumento incontrolado de respuesta Th1, aumento de la síntesis
de INF-gamma y ausencia de células NKT, aumento de
proliferación de linfocitos T CD8
• Diagnóstico: mutación en SH2D1A/SAP (SH2 domain-
containing gene 1ª/SLAM-associated protein) que se encuentra
en el 50-70% de los pacientes.
XIAP en un numero menor de casos. Atención ITK en niñas¿Existe tratamiento para esta
IDP?¿Existe tratamiento para esta IDP? 1. No existe tratamiento 2. Tratamiento antiviral 3. GGIV periódica 4. Trasplante de precursores hematopoyéticos
¿Existe tratamiento para esta IDP? 1. No existe tratamiento 2. Tratamiento antiviral 3. GGIV periódica 4. Trasplante de precursores hematopoyéticos
CASO CLINICO 3
CASO 3
Lactante de 8 meses que consulta por irritabilidad y deterioro
nivel consciencia
• No antecedentes familiares ni personales de interés
• Febril las 24 h previas
• Se decide ingreso para estudio:
- TC cerebral normal
- LCR: leucocitos: 61 céls/mm3 (60% linf)
glucosa: 66 mg/dl
proteínas: 0.63 g/l
• OD: encefalitis-meningitis infecciosa
• Se inicia tratamiento empírico con cefotaxima y aciclovir IV
• Resultados microbiológicos: PCR positiva VHS-1CASO 3
• Buena evolución clínica. Dado de alta con mínimas
secuelas (tratamiento anticomicial)
• A los 12 meses de edad reingresa por un cuadro
similar de deterioro neurológico nuevo
episodio de encefalitis VHS -1¿Consideran adecuado llevar
a cabo un estudio inmunitario
en este momento?¿Consideran adecuado llevar a cabo un
estudio inmunitario en este momento?
1. No, no cumple criterios para indicarlo.
2. Si, ha presentado una infección grave y
recurrente.
3. No, porque son por el mismo microorganismo y
estaría indicado sólo si fuesen diferentes
4. Sí, ya que una infección del SNC siempre tiene
que estudiarse desde el punto de vista
inmunológico.¿Consideran adecuado llevar a cabo un
estudio inmunitario en este momento?
1. No, no cumple criterios para indicarlo.
2. Si, ha presentado una infección grave y
recurrente..
recurrente
3. No, porque son por el mismo microorganismo y
estaría indicado sólo si fuesen diferentes
4. Sí, ya que una infección del SNC siempre tiene
que estudiarse desde el punto de vista
inmunológico.¿Qué estudios solicitarían?
¿ Qué estudios solicitarían? 1. Hemograma e inmunoglobulinas. 2. Test de oxidación. 3. Hemograma, inmunoglobulinas, inmunofenotipo linfocitario, función linfocitaria, serología frente a VIH. 4. Respuesta 3 + estudio de la vía de TLR-3.
¿ Qué estudios solicitarían?
1. Hemograma e inmunoglobulinas séricas.
2. Test de oxidación.
3. Hemograma, inmunoglobulinas,
inmunofenotipo linfocitario, función linfocitaria,
serología frente a VIH.
4. Respuesta 3 + estudio de la vía de TLR
TLR--3.Déficit de TLR 3
Susceptibilidad aumentada a
encefalitis ¿recurrente? por
VHSINMUNIDAD INNATA: TLR 3
Genetic deficiencies of innate immune signalling in human infectious disease.
Lancet Infect Dis. 2009 ;9:688-98.Genetic deficiencies of innate immune signalling in human infectious disease.
Lancet Infect Dis. 2009 ;9:688-98.TLR (Toll like receptor)
• Toll like receptors (TLR) son proteínas transmembrana que actúan
como mediadores intracelulares liberando una serie de citocinas
(interferón tipo I) en respuesta al reconocimiento de partículas
microbianas (PAMPs).
• Los TLRs son activados por PAMPs específicos y muy importantes
en la inmunidad innata
• IDP con defectos primarios de la señalización TLR:
- déficit IRAK-4 / MyD88
- déficit TLR3, UNC-93B,….Déficit de TLR 3
• Incidencia: Se desconoce. Descrita por primera vez en 2007.
• Manifestaciones clínicas: episodios ¿recurrentes? de meningo-
encefalitis por VHS-1. ¿VHS-2?
• Diagnóstico: sospechar alteración de la vía innata ante cuadros
infecciosos con pocas manifestaciones de tipo inflamatorio (mínima
elevación de PCR y leucocitos…). De todas maneras, UNA SOLA
INFECCIÓN GRAVE DEL SNC POR VHS, PODRÍA DEBERSE A UNA
IDP (ORIGEN CONOCIDO O DESCONOCIDO…).
• Patogenia: deterioro en la inducción de INF-α/β/γ frente a las partículas
virales
TLR3 deficiency in patients with herpes simplex encephalitis.
Science 2007 14;317:1522-7.¿Existe tratamiento para esta
IDP?¿Existe tratamiento para esta IDP? 1. No existe tratamiento 2. Trasplante de precursores hematopoyéticos 3. Profilaxis con aciclovir +/- IFN-α /INF β 4. Terapia génica
¿Existe tratamiento para esta IDP?
1. No existe tratamiento
2. Trasplante de precursores hematopoyéticos
3. Profilaxis con aciclovir +/- IFN--α /INF β
+/- IFN
4. Terapia génica
muy discutido!!!CASO CLINICO 4
CASO 4
Niña de 7 años remitida para estudio por infecciones bacterianas
de repetición e infecciones virales cutáneas
• AF: Consanguinidad
• AP: -Dermatitis atópica importante desde los primeros meses
de vida (sobreinfección cutánea por S.aureus).
-Alergia pescado
-Molluscum contagiosum muy extenso
-Herpes zóster recurrente
-Otitis media supurada de repetición
-Cuadros de sobreinfección respiratoria. 3 Neumonías
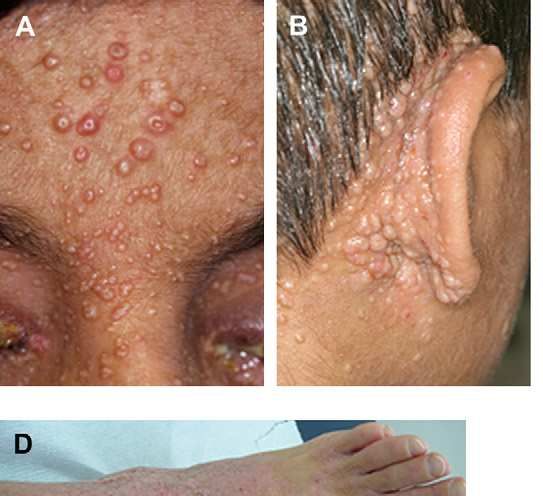
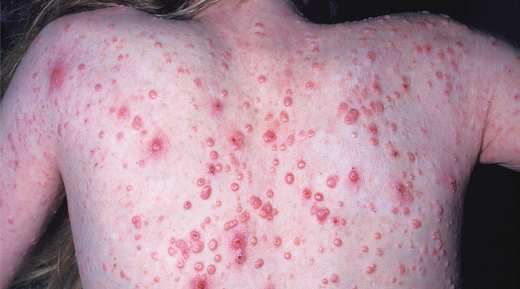
-Retraso pondero – estatural.Molluscum contagiosum en espalda y lesiones de DA sobreinfectada
¿Consideran adecuado llevar
a cabo un estudio inmunitario
en este momento?¿Consideran adecuado llevar a cabo un estudio inmunitario en este momento? 1. No, no cumple criterios para indicarlo. 2. Sí, la infección por molluscum siempre indica una IDP 3. No, se trata de un paciente atópico mal controlado. 4. Sí, se trata de una entidad potencialmente grave.
¿Consideran adecuado llevar a cabo un estudio inmunitario en este momento? 1. No, no cumple criterios para indicarlo. 2. Sí, la infección por molluscum siempre indica una IDP 3. No, se trata de un paciente atópico mal controlado. 4. Sí, se trata de una entidad potencialmente grave.. grave
CASO 4
Niña de 7 años remitida para estudio por infecciones bacterianas
de repetición en infecciones virales cutáneas
EC: Linfopenia.
Disminución de linf T CD4 y CD8.
Leve disminución de céls B. NK normales
Alteración de la respuesta a mitógenos
Ig G e Ig A normales. Ig M disminuida. Ig E 5300
Eosinofilia (1700 céls/ml)
Correcta respuesta vacunal (excepto a tétanos)¿Cuál sería su diagnóstico en
este momento?¿Cuál sería su diagnóstico en este momento? 1. Evidentemente se trata de una infección por VIH. 2. Se confirma que se trata de un paciente atópico mal controlado. 3. Síndrome de hiperIgE AD (déficit de STAT-3) 4. Inmunodeficiencia combinada (leaky-SCID)
¿Cuál sería su diagnóstico en este momento?
1. Evidentemente se trata de una infección por
VIH
2. Se confirma que se trata de un paciente
atópico mal controlado
3. Síndrome de hiperIgE AD (déficit de STAT-3)
4. Inmunodeficiencia combinada (leaky-
leaky-SCID)CASO 4
Infecciones vía aérea sup e inf: S. pneumoniae,Hib…
Infección cutánea viral y bacteriana (S aureus)
Fenomenos alérgicos, atopia
Aumento Ig E y eosinofilia
Mutación en DOCK8CASO 4 Noviembre 2009
CASO 4
Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-
recessive form of hyper-IgE syndrome.
JACI 2009;6: 1289-1302.DOCK8
• Incidencia: IDP infrecuente. Herencia AR (se han encontrado mutaciones
homocigotas en DOCK8 en pacientes con HIES-AR).
• Manifestaciones clínicas (similares a las de pacientes con formas “atípicas”
de HIES): Infecciones respiratorias recurrentes. Infecciones cutáneas extensas
por virus y bacterias. Atopia grave. Susceptibilidad a neoplasias.
• Diagnóstico: Mutación en el gen citoquinesis 8 (DOCK8)
• Patogenia: DOCK8 pertenece a una superfamilia (DOCK180) de factores de “in-
intercambio” y se encarga de regular reordenamientos de componentes del
citoesqueleto necesarios para la estructura celular, migración, adhesión …
• Tratamiento: GG ev de manera periódica. Tratamiento de los procesos
infecciosos. TPH
Combined immnodeficiency associated with DOCK8 mutations
NEJM 2009:2046-55.CASO CLINICO 5
CASO 5
Niño de 11 años, diagnosticado de enfermedad de Bruton
(agammaglobulinemia ligada al cromosoma X) y que
remiten para estudio por alteración neurológica
• AP: diagnosticado a los 6 años de edad (antecedente de
infecciones bacterianas, varias neumonías, otitis) detectando
hipogammaglobulinemia con ausencia de linfocitos B
El paciente no ha recibido tratamiento porque la familia se
negaba y no creía el diagnóstico
• ENFERMEDAD ACTUAL:
La familia refiere que desde hace 3-4 semanas el paciente ha
presentado un cambio carácter marcado, con tendencia al sueño
y febrícula intermitente. También presenta cefalea¿En que posibilidades diagnósticas pensarían?
¿ En que posibilidades diagnósticas pensarían? 1. Solo puede ser un tumor del SCN 2. De momento no hay que estudiarlo. Parece psicosomático y tenderá a resolverse 3. Puede ser una infección del SNC pero no tiene relación con su enfermedad de base 4. Puede ser una infección del SNC y relacionarse con una complicación de su enfermedad de base
¿ En que posibilidades diagnósticas pensarían? 1. Solo puede ser un tumor del SCN 2. De momento no hay que estudiarlo. Parece psicosomático y tenderá a resolverse 3. Puede ser una infección del SNC pero no tiene relación con su enfermedad de base 4. Puede ser una infección del SNC y relacionarse con una complicación de su enfermedad de base
CASO 5
Niño de 11 años, diagnosticado de enfermedad de Bruton
(agammaglobulinemia ligada al cromosoma X (ALX) y que
remiten para estudio por alteración neurológica
• EC:
TC craneal: normal
LCR: compatible con meningitis viral. PCR positiva a enterovirus
RM: lesiones compatibles con encefalitis
MENINGOENCEFALITIS POR
ENTEROVIRUS¿Hay algún factor de riesgo añadido
a la enfermedad de base de este
paciente para predisponerlo a esta
infección ?¿ Hay algún factor de riesgo añadido a la
enfermedad de base de este paciente para
predisponerlo a esta infección ?
1. No, la enfermedad de Bruton solo predispone a
infecciones bacterianas
2. No, ha tenido mala suerte
3. Si, porque no estaba con tratamiento con gammaglobulina
sustitutiva
4. Si, ya que tiene 11 años y el riesgo aumenta con la edad¿ Hay algún factor de riesgo añadido a la
enfermedad de base de este paciente para
predisponerlo a esta infección ?
1. No, la enfermedad de Bruton solo predispone a
infecciones bacterianas
2. No, ha tenido mala suerte
3. Si, porque no estaba con tratamiento con gammaglobulina
sustitutiva
4. Si, ya que tiene 11 años y el riesgo aumenta con la edadALX y encefalitis por enterovirus
ALX y encefalitis
¿Cuál es el tratamiento de la agammaglobulinemia ligada a X?
¿Cuál es el tratamiento de la
agammaglobulinemia ligada a X?
1. Trasplante de precursores hematopoyéticos
2. Profilaxis antibiótica
3. Tratamiento sustitutivo con gammaglobulina
4. Terapia génica¿Cuál es el tratamiento de la
agammaglobulinemia ligada a X?
1. Trasplante de precursores hematopoyéticos
2. Profilaxis antibiótica
3. Tratamiento sustitutivo con gammaglobulina
4. Terapia génicaALX • Déficit producción de Inmunoglobulinas por ausencia de linfocito B • Clínica: infec bacterianas pero posibilidad de infec virales
DIAGNÓSTICO DE ALX
(www.esid.org)
Definitivo
Paciente masculino con < 2% linfocitos B CD19+ y al menos uno de los siguientes criterios:
1)Mutación del gen Btk
2) Ausencia de RNA m BtK en neutrófilos o monocitos (northern blot)
3) Ausencia de la proteina Btk en monocitos o plaquetas
4) Primos por rama materna, tíos con < 2% linfocitos B CD19+
Probable
Paciente masculino con < 2% linfocitos B CD19+ y que cumple todos los siguientes criterios:
1) Inicio de infecciones bacterianas de repetición en los primeros 5 años de vida.
2) Niveles de Ig G, Ig M e Ig A por debajo de 2 DS según los valores normales para la edad
3) Ausencia de isohemaglutininas y/o pobre respuesta a vacunas.
4) Exclusión de otras causas de hipogammaglobulinemia.
Posible
Paciente masculino con < 2% linfocitos B CD19+ en el cual se han descartado otras causas de
hipogammaglobulinemia y al menos con uno de los siguientes criterios:
1) Inicio de infecciones bacterianas de repetición en los primeros 5 años de vida.
2) Niveles de Ig G, Ig M e Ig A por debajo de 2 DS según los valores normales para la edad
3) Ausencia de isohemaglutininasCONCLUSIONES
• Es necesario pensar en las IDP para poder llegar a diagnosticarlas
• La identificación de las bases genéticas de las IDP han facilitado un
mejor diagnóstico y mayores opciones terapéuticas
• Un diagnostico precoz mejora el pronóstico de estos pacientes
• A pesar de todo, muchos pacientes siguen sin estar diagnosticados
debido a que no se “reconocen” o no existen las técnicas
diagnósticas adecuadas.MUCHAS GRACIAS POR SU
ATENCIÓNTambién puede leer

















































