Efecto del dicloroacetato (DCA) y de inhibidores de tirosín quinasas sobre células tumorales con mutaciones en el DNA mitocondrial - TRABAJO DE ...
←
→
Transcripción del contenido de la página
Si su navegador no muestra la página correctamente, lea el contenido de la página a continuación
Efecto del dicloroacetato (DCA) y de inhibidores
de tirosín quinasas sobre células tumorales con
mutaciones en el DNA mitocondrial
ión por integrinas
e las integrinas a la
lugar a su oligo-
y a la activación de
autofosforilación. La
Src produce nuevas
nes en FAK que dan
clutamiento via SH2
s proteínas de
n
TRABAJO DE FIN DE GRADO
Autor: Fernando de la Figuera (4º Biotecnología)
Directores: Ruth Soler y Alberto Anel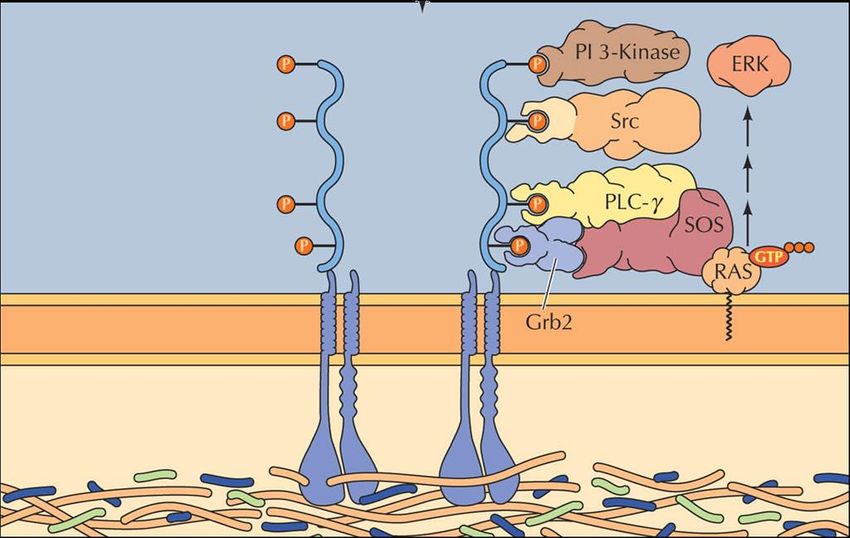
ÍNDICE
Resumen / Abstract
1. Introducción: bases moleculares del cáncer
1.1 El Cáncer o transformación tumoral
1.2 Hallmarks o marcadores del cáncer
1.3 Trasformación metastásica
1.4 El metabolismo tumoral; efecto Warburg
1.5 Terapias antimetabólicas: dicloroacetato (DCA)
1.6 Las fosforilaciones como reguladores de la señalización celular:
receptores tirosín-quinasas y vías de señalización asociadas
1.7 La quinasa de adhesión focal
1.7.1 El gen PTK2 y la regulación de la expresión de FAK
1.7.2 La proteína FAK
1.7.3 El papel principal de FAK: vía dependiente de quinasas
y señalización mediada por integrinas
1.7.4 El rol de FAK en la anoikis
1.7.5 FAK en el núcleo: vía independiente de quinasas
2. Antecedentes y estado actual del tema
3. Objetivos
4. Materiales y métodos
4.1 Cultivos celulares
4.2 Generación de cíbridos transmitocondriales
4.3 Experimentos con inhibidores
4.3.1 Estudio del inhibidor general de PTKs genisteína
4.3.2 Efecto del defactinib en el modelo celular L929
4.3.3 Efecto de la terapia combinada: defactinib y DCA
4.3.4 Ensayos de la inhibición del crecimiento
mediante la técnica de MTT
4.4 Análisis de la expresión de proteínas
mediante Western Blot
4.5 Estudio de la localización de FAK mediante
microscopía de fluorescencia
4.6 Análisis estadístico
5. Resultados y discusión
5.1 Fosforilación en tirosina de sustratos celulares
5.1.1 Estudio de la expresión de PTK por Western Blot
5.1.2 Inhibición de las PTKs con genisteína
5.2 Niveles de expresión de FAK y FAK fosforilada en Y397
5.3 Experimentos de citotoxicidad
5.4 Estudio de la localización de FAK mediante
microscopía de fluorescencia
5.3.1 Muerte celular
5.3.2 Inhibición del crecimiento
5.3.3 Efecto combinado del defactinib y el DCA
6. Conclusiones / Conclusions
7. Bibliografía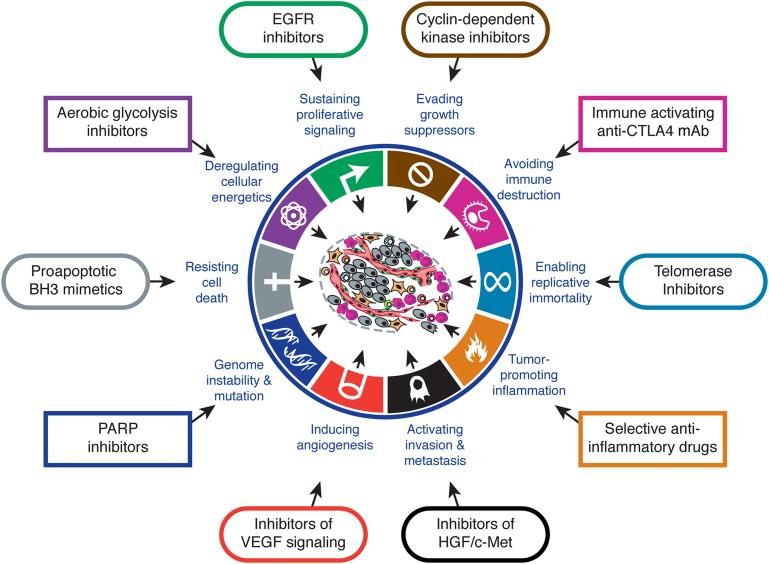
Resumen
En trabajos anteriores se había demostrado mediante el estudio de su
quinoma que la línea celular L929dt, la cual se generó de forma espontánea en
nuestro laboratorio hace años, tenía una actividad exacerbada de las actividades
proteín tirosín quinasa (PTK) respecto a sus parentales L929 (fibroblastos de ratón).
Las células L929dt, a diferencia de las células L929 parentales, exhiben mutaciones
en el DNA mitocondrial, crecen en suspensión y tienen un metabolismo de tipo
fermentativo. La dependencia de la actividad PTK en este modelo in vitro de células
metastásicas (L929dt) se demostró por su sensibilidad a la genisteína, un inhibidor
general de PTK. Uno de los sustratos que exhibían una mayor diferencia en su
fosforilación entre las células L929 y L929dt era la quinasa de adhesión focal (FAK).
En este trabajo se ha estudiado si su inhibición farmacológica con defactinib podría
afectar de forma diferencial a las células L929dt. Los resultados obtenidos
demuestran la localización nuclear de FAK en las células L929dt y sugieren que la
inhibición de la actividad quinasa de FAK no es especialmente relevante para
eliminar estas células, ya sea como monoterapia o en combinación con el fármaco
antimetabólico dicloroacetato (DCA).
Abstract
Previously by studying their kinome, it was proven that L929dt cells, a cell line
that appeared spontaneously in our lab, had a high protein tyrosine kinase (PTK)
activity compared to L929 parental cells. L929dt cells, unlike the original L929 cells,
have mitochondrial DNA mutations, they growth detached and they have a
fermentative-type metabolism. L929dt cells were proven to be dependent of PTK
activity by the use of genisteine, a general PTK inhibitor. One of the substrates which
had a meaningful difference in his fosforilation between L929 and L929dt cells was
the focal adhesión kinase (FAK). In this project, we studied if the use of defactinib
(FAK pharmacological inhibitor) could selectively affect L929dt cells. The results
obtained proved that FAK was localized in the nucleus and suggested that FAK
inhibition it is not really relevant neither with defactinib nor with the antimetabolic
drug dichloroacetate, or the combination of both.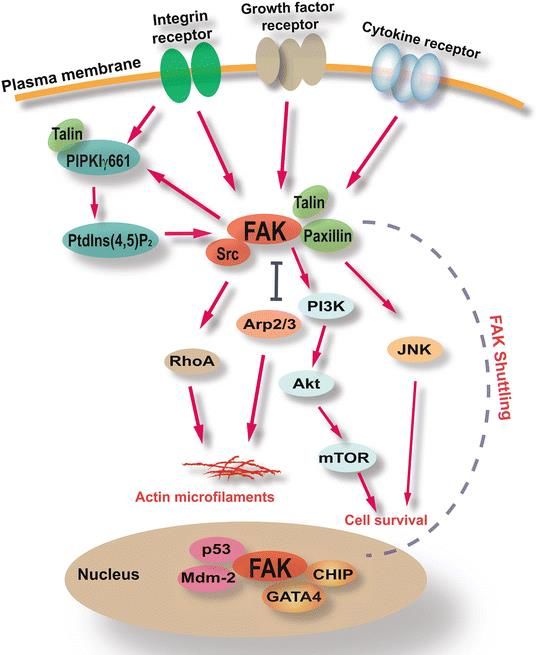
1. Introducción: bases moleculares del cáncer
1.1 El Cáncer o transformación tumoral
El cáncer es una enfermedad que se caracteriza por la presencia de un fenómeno
conocido como transformación celular. Este fenómeno lleva a la proliferación anormal e
incontrolada de las células y puede llevar a la muerte si no se trata a tiempo. Para que haya
cáncer se requieren al menos tres “requerimientos” genéticos1:
1. La desregulación del ciclo celular (entrada en ciclo constante).
2. Una capacidad replicativa sin límites por el mantenimiento infinito de los telómeros.
3. La inactivación de mecanismos que aseguran la integridad del genoma (evasión de la
muerte celular programada y acumulación de mutaciones).
Estas alteraciones genéticas llevan a la aparición de un tumor. Si este continúa su
desarrollo podría adquirir la capacidad de invadir otros tejidos, un proceso denominado
metástasis2. Tras décadas de investigación se han ido describiendo detalladamente las
diferentes características de esta enfermedad3 y junto a ello decenas de biomarcadores
concretos. En consecuencia, se han desarrollado fármacos antitumorales más específicos y
con mayor efectividad.
Figura 1. Hallmarks
del cáncer y terapias
propuestas para cada
uno de ellos.3
1.2 Hallmarks o marcadores en el cáncer
Se han caracterizado numerosos biomarcadores moleculares3 para el diagnóstico y
el tratamiento del cáncer, los siguientes son algunos de los más relevantes:
Página 1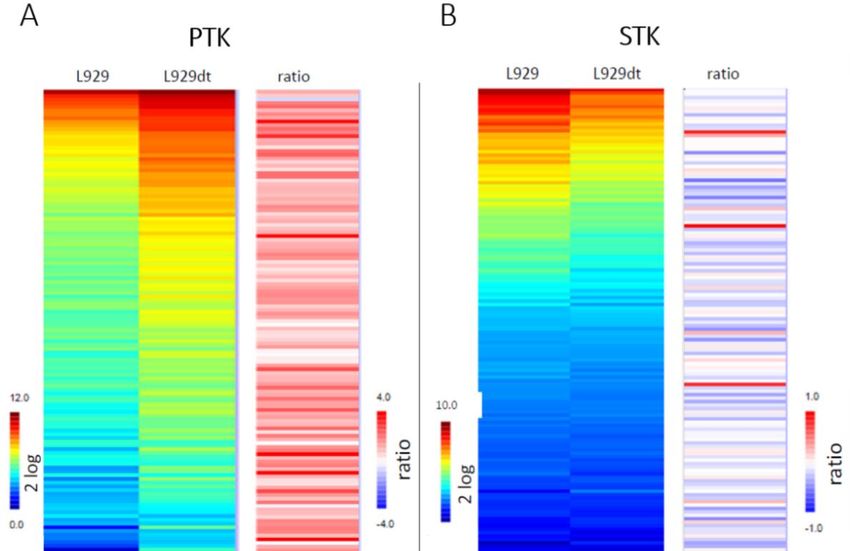
• Potencial replicativo ilimitado: se observa una mayor expresión de la telomerasa; la DNA
polimerasa que replica los telómeros del cromosoma.
• Oncogenes: genes que al activarse (ganancia de función) favorecen la transformación
tumoral4. El crecimiento celular descontrolado puede deberse a desregulaciones o
alteraciones en rutas de supervivencia (Ras/Erk etc.), y de proliferación (PI3K/Akt, etc.),
entre otras. Por ejemplo, las quinasas involucradas en estas rutas (SRC), los propios
receptores que las desencadenan (EGFR), así como ciertos factores de transcripción (c-
Myc) pueden estar sobreexpresados, mutados y/o sobreactivados. Por todo ello, se les
conoce como oncogenes.
• Resistencia a la muerte celular programada: este fenómeno se debe a múltiples
factores5. Los más importantes son la pérdida de función en genes o proteínas supresoras
de tumores (p53, Rb…), o la activación y/o sobreexpresión de proteínas anti-apoptóticas
cómo Bcl-2, Bcl-XL y Mcl-1.
• Remodelación del citoesqueleto: la pérdida de cadherinas y de uniones intercelulares en
general, son considerados como marcadores de proceso de metástasis6. Esta característica
permite a las células tumorales “desprenderse” del tumor primario, circular por el torrente
sanguíneo y linfático, y finalmente colonizar otro tejido2,6.
• Angiogénesis prolongada: es la elongación o remodelación de vasos sanguíneos
preexistentes. Suele ser una consecuencia de la hipoxia y de la señalización oncogénica,
las cuales modulan la expresión de proteínas7 como el factor de crecimiento endotelial
vascular (VEGF) y algunos miembros pertenecientes a la familia de los factores de
crecimiento de fibroblastos (FGF). Las células endoteliales reclutadas secretan
8
metaloproteasas que degradan la matriz extracelular y angiopoyetinas que reclutan a los
pericitos9. Los pericitos son células que pueden diferenciarse a células endoteliales o
musculares lisas10 y son características de un vaso sanguíneo maduro.
• Evasión de la respuesta inmunitaria: algunos mecanismos que pueden incidir en esta
evasión son el aumento de la secreción de citoquinas inhibidoras de células del sistema
inmune, la expresión de PD-L1 y CTLA411 o la pérdida del MHC-I12, el complejo de
presentación de antígenos a los linfocitos CD8+.
• Remodelación del metabolismo energético: en las células tumorales se observa el efecto
Warburg; una preferencia por la fermentación de la glucosa incluso en presencia de
oxígeno13,14.
Página 2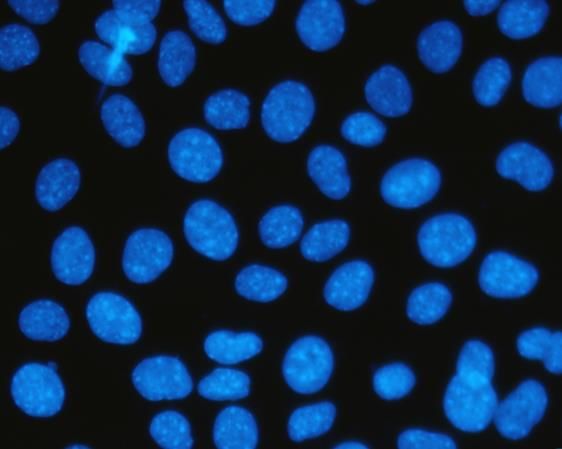
1.3 Transformación metastásica
Durante el proceso de transformación metastásica las células tumorales evolucionan
a partir de un fenotipo de tipo epitelial hacia uno de tipo mesenquimal2,6.
Esto sucede habitualmente por el cambio progresivo de proteínas de membrana y la
reorganización de proteínas del citoesqueleto que resulta en una pérdida de adherencia de
las células tumorales a su matriz extracelular, adquiriendo la capacidad de migrar. El
proceso de metástasis puede clasificarse en 5 fases principales2: invasión del tejido
primario, intravasación, circulación por el torrente sanguíneo, extravasación y colonización
del nuevo tejido.
Una vez las células tumorales han alcanzado un nuevo tejido, se produce una
reversión de vuelta al fenotipo epitelial, por lo que se vuelven a adherir al tejido formando
micrometástasis. Seguidamente, aquellas células que sigan proliferando formaran
metástasis en sensu stricto, el cual se asocia con un peor pronóstico y una mayor
mortalidad.
1.4 El metabolismo tumoral; efecto Warburg
Otto Warburg postuló como hipótesis que la causa principal del cáncer son los
defectos mitocondriales que impiden realizar correctamente la respiración celular9. Se
define efecto Warburg como la preferencia de algunas células por la fermentación de la
glucosa, incluso en presencia de oxígeno14,15, a pesar de ser energéticamente menos
rentable comparado con la vía anaerobia. Este fenómeno se ha observado en diferentes
tipos de cáncer aunque actualmente se sabe que no todas las células tumorales presentan
fallos en las enzimas necesarias para la respiración aerobia14,15 y se ha relacionado con
otras causas como la hipoxia , los oncogenes, la pérdida de supresores de tumores etc.
La hipoxia se suele dar en las primeras etapas de diversos cánceres por falta de
irrigación sanguínea7,16. En estos casos el factor inducible por hipoxia (HIF-1α) induce la
expresión de VEGF (entre otros factores de crecimiento que promueven la angiogénesis) o
de la Piruvato Deshidrogenasa Quinasa (PDK)17, una enzima que promueve
preferentemente la glicólisis anaerobia e inhibe el sistema OXPHOS mitocondrial.
Los oncogenes activan genes de transportadores y enzimas que promueven la
glicólisis aerobia y el anabolismo a partir de los metabolitos del ciclo de Krebs mientras que
los supresores de tumores intentan inhibir estos efectos y favorecer de diferentes maneras
el sistema OXPHOS18, 19.
Página 3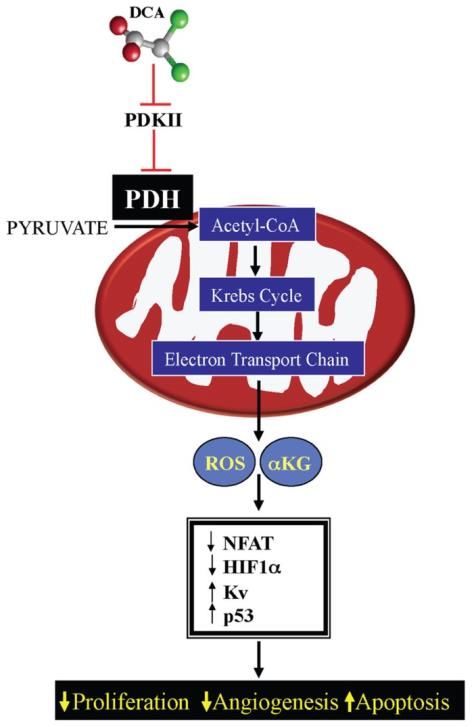
Se han descrito posibles consecuencias que tendría este efecto para favorecer a las
células tumorales a proliferar y sobrevivir14, 17.
En primer lugar, les permitiría generar potencial reductor en forma de NADPH (a
partir de la ruta de las pentosas fosfato) y ATP (a partir de la glucólisis anaerobia), para
poder cubrir las elevadas exigencias energéticas de forma más rápida.
En segundo lugar, se generaría un microambiente más ácido, por el exceso de
producción de lactato, que favorecería la evasión de la respuesta inmune. Así mismo, un
cambio en las rutas de señalización asociadas al fenotipo glucolítico podría aumentar la
producción de especies reactivas de oxígeno (ROS), que podrían provocar daños tanto en
el DNA mitocondrial como en el nuclear, aumentando así la inestabilidad genética y la
acumulación de nuevas mutaciones.
1.5 Terapias anti-metabólicas: dicloroacetato (DCA)
El dicloroacetato o DCA es una molécula soluble de 150 Da que ha sido utilizada en
la clínica para el tratamiento de la acidosis láctica20,21. Más recientemente, algunos
trabajos realizados con modelos preclínicos, entre los cuales se encuentra nuestro
grupo de investigación22, han demostrado que el DCA también posee una capacidad
antitumoral23,24.
Su principal diana es la piruvato
deshidrogenasa quinasa (PDK) responsable de
inhibir a la piruvato deshidrogenasa (PDH),
involucrada en la conversión del piruvato a acetil-
CoA destinado al ciclo de Krebs.
Es por ello que, el uso del DCA en la
oncología podría presentarse como una estrategia
para el tratamiento de tumores con un efecto
Warburg acusado. Por lo tanto, favorecer la
respiración por el aumento de la actividad de la PDH
permitiría: reducir la acidez del microambiente
tumoral, potenciar la respuesta inmunitaria, reducir
la angiogénesis y el comportamiento tumoral
asociado al fenotipo de tipo glucolítico.
Figura 2. Mecanismo de acción del DCA24.
Página 4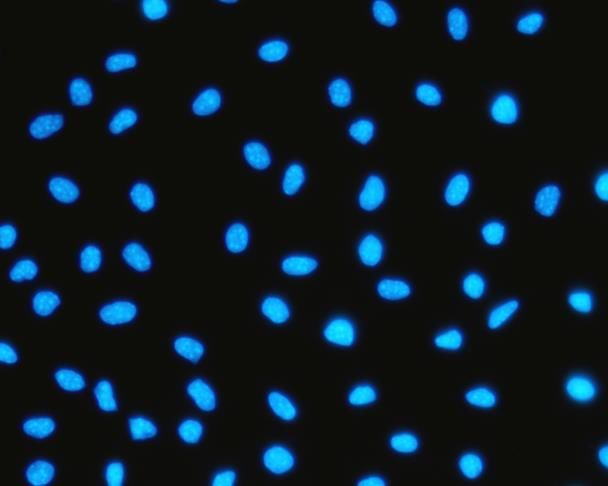
1.6 La fosforilación como reguladora de la señalización celular: receptores
tirosín-quinasas y vías de señalización asociadas
Los receptores tirosín-quinasas (RTKs) son proteínas transmembrana que
responden a estímulos exteriores (ligandos que inducen su dimerización) y desencadenan
la activación de un gran número de vías de señalización (asociadas a procesos
proliferativos, de supervivencia, del ciclo celular, apoptosis, etc.) mediante cascadas de
fosforilaciones20 iniciadas a través de su dominio quinasa citoplásmico.
Figura 3. Mecanismo general de acción de los RTKs25.
Dentro de esta extensa familia se encuentran los receptores de factores de crecimiento
(EGFR, PDGFr, FGFr…)26 o los receptores de las integrinas responsables de reclutar, por
ejemplo, a la quinasa de adhesión focal (FAK)27. Tanto los RTKs como muchas de las
proteínas que se reclutan a partir de las vías conectadas directamente (MAPK, PI3K/Akt…)
o los factores de transcripción, que se fosforilan por esas rutas y se translocan al núcleo,
son considerados actualmente como posibles oncogenes.
1.7 La quinasa de adhesión focal
1.7.1 El gen PTK2 y la regulación de la expresión de FAK
El gen PTK2 (localización: 8q24.3)28 es el gen que codifica para la quinasa de
adhesión focal (FAK). Se trata de un gen del que se conocen numerosas mutaciones y
Página 5variantes generadas por ayuste o splicing alternativo. Estas alteraciones de PTK2, que
suelen acortar la proteína, se encuentran en su mayoría en patologías tumorales y se han
asociado a un peor pronóstico clínico28-30. El aumento de su expresión, la pérdida de
dominios de regulación de la proteína por splicing alternativo o los cientos de polimorfismos
del gen encontrados en diversos tipos de cáncer, contribuyen a una mayor actividad global
de la proteína. En estos casos, la agresividad tumoral se vería aumentada porque, aunque
FAK no es considerado un oncogén, se encuentra involucrada en diversas vías de
proliferación, movilidad y crecimiento. Por lo tanto, una mayor expresión de FAK y la
existencia de polimorfismos o deleciones en el gen PTK2 han sido identificados como
biomarcadores claves en el cáncer urotelial, de pulmón, de mama o de tiroides31-34.
Un ejemplo de splicing alternativo es la pérdida del exón 33 en pacientes graves de
cáncer de tiroides y de mama27, 31, el cual se asocia a un peor pronóstico en los pacientes y
a la elevada expresión de FAK en esos tipos de cáncer31,32.
La expresión del ARN mensajero (ARNm) originado a partir de PTK2 es similar en
todos los tejidos32 y está regulada principalmente por el factor nuclear potenciador de las
cadenas ligeras kappa de las células B activadas (NF-ΚB)27 . La traslocación al núcleo de
NF-ΚB está a su vez regulada por factores de crecimiento, receptores de citoquinas
proinflamatorias o receptores de integrinas, las cuales pueden fosforilar a NF-ΚB. Además
de NF-ΚB existen otros reguladores que favorecen la expresión de PTK2 como AGO2,
NANOG o PEA3 que se han encontrado en diversas patologías tumorales27.
Por otra parte, se ha descrito
que p53 es un inhibidor de la expresión
de PTK2. El equilibrio entre PTK2, FAK
y p53 suele verse desplazado hacia la
degradación de p53 en las etapas más
avanzadas del cáncer, habitualmente
como consecuencia de la pérdida de
adherencia observados durante el
proceso de metástasis y la migración de
FAK al núcleo35.
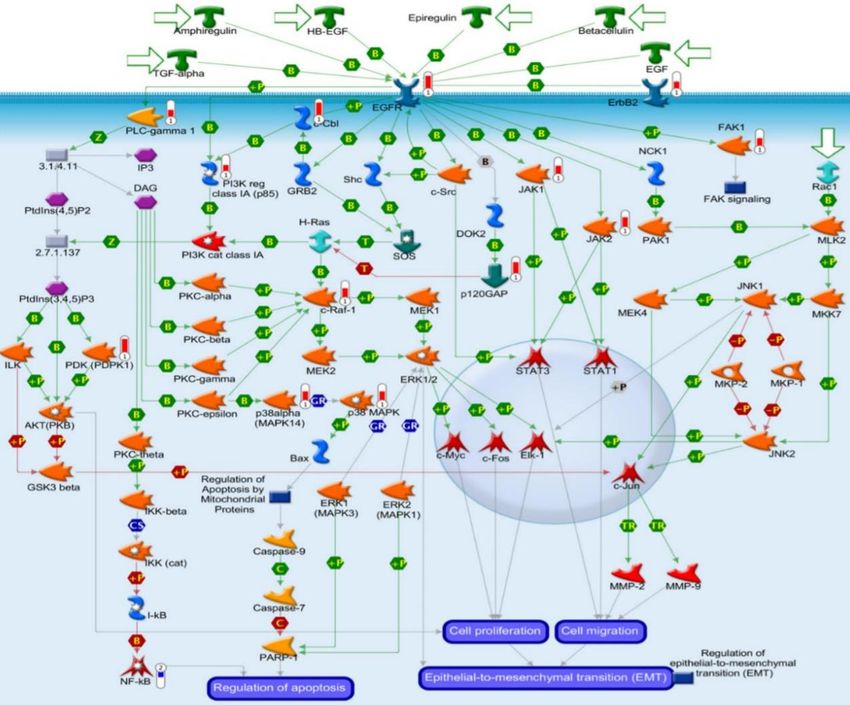
Figura 4. Algunas rutas de señalización
asociadas a FAK36. En el esquema se
puede observar el doble papel de FAK.
Página 61.7.2 La proteína FAK
FAK es una proteína de 125 kDa citosólica con capacidad de traslocarse al núcleo.
Es clave en la señalización por integrinas y otros receptores27,35,36 . Las integrinas y los
receptores de citoquinas o diversos factores de crecimiento pueden dimerizar y reclutar a
FAK27. El reclutamiento de FAK induce su autofosforilación en la posición aminoacídica
Y397, la cual es la responsable de reclutar a la proteína SRC37.
La proteína SRC fosforila a su vez a FAK en varios residuos permitiendo la
interacción con otras quinasas que, a su vez, regulan numerosas vías de señalización de
proliferación y supervivencia (PI3K/AKT, MAPK, etc.). Esta vía de señalización mediada por
FAK se conoce como dependiente de quinasas27. Así mismo, se ha descrito que la pérdida
de adherencia y/o el estrés oxidativo favorecen la traslocación de FAK al núcleo por una vía
independiente de quinasas35. Esta vía tiene como consecuencia la inhibición de P53, entre
otras alteraciones27,35.
1.7.3 El papel principal de FAK: vía dependiente de quinasas y señalización
mediada por integrinas.
En el caso que las integrinas estén unidas a la matriz extracelular, oligomerizan y reclutan a
FAK en las zonas de adhesión focal. Esto desencadena27 la activación de rutas celulares de
remodelación del citoesqueleto, proliferación, inhibición de la apoptosis y supervivencia.
Una de las dianas de estas rutas es NF-ΚB, ergo, aumenta la expresión de PTK2 y se
produce un bucle de retroalimentación positiva. La señalización por integrinas es además
una de las formas de regular la anoikis38 (detallado en el apartado 1.7.4)
1.7.4 El rol de FAK dentro de la muerte celular programada por anoikis
La anoikis es un subtipo de muerte celular programada involucrada en el proceso de
metástasis y se desencadena, entre otros motivos, si FAK no está señalizando en las
integrinas. Por una parte, como se citaba en el apartado 1.3, en el proceso de metástasis
las células tumorales evolucionan de un fenotipo de tipo epitelial hacia uno de tipo
mesenquimal6.. Esto requiere que las células pierdan la adherencia y extravasen a otros
tejidos, de modo que la anoikis es un mecanismo celular que podría bloquear la metástasis
en los casos en los que las células han perdido la interacción FAK-Integrinas-Matriz
extracelular38. Por otra parte, como se comentaba en el apartado 1.2, una de las
características del cáncer es la resistencia a la muerte celular programada. Dentro de esos
mecanismos, uno de ellos es lograr la resistencia a anoikis3,38.
Página 7En consecuencia, FAK tiene un papel clave a la hora de regular el mantenimiento de
la adherencia celular pero las células tumorales metastásicas logran evadir esta regulación.
1.7.5 FAK en el núcleo: vía independiente de quinasas
Se ha descrito que la pérdida de adherencia o el estrés oxidativo aumentan la
cantidad de FAK citosólica libre, por lo que FAK puede importarse al núcleo con ayuda de la
proteína antiapoptótica XIAP35. Esta traslocación también se ha visto que ocurre
empíricamente si la actividad quinasa de la proteína se encuentra inhibida. Esta vía
favorece la ubiquitinación y degradación de p53, también conocido como “guardián del
genoma” 19.
2. Antecedentes y estado actual del tema
En estudios realizados por nuestro grupo de investigación12 se caracterizó una línea
celular derivada de la línea fibroblástica de ratón L929, que se denominó L929dt, la cual
había perdido la adherencia al frasco de cultivo. Además se demostró que las células
L929dt habían perdido la expresión del MHC-I, el complejo de presentación de antígenos a
los linfocitos CD8+, primordial en la respuesta inmunitaria frente a tumores. Un estudio más
detallado22 reveló que estas células habían adquirido mutaciones en el DNA mitocondrial,
concretamente, en la subunidad ND2 del complejo I respiratorio, viéndose significativamente
afectada su capacidad de generar energía a través de la vía aerobia. Como consecuencia
de su disfunción mitocondrial, se demostró que estas células se encuentran forzadas a
depender de la glucólisis anaerobia, por lo que presentan un efecto Warburg muy
acusado14,15.
Además, se demostró que el fenotipo de estas células se debe a las mutaciones en
su mtDNA, ya que cíbridos que expresan el mtDNA de las células L929dt en el fondo
nuclear de las células L929 (L929dt) se comportan como L929dt. Se demostró además que
la línea L929dt había adquirido un comportamiento especialmente agresivo y metastásico in
vivo. Para poder comprender las diferencias entre las células L929 y las L929dt se realizó
un análisis comparado del quinoma en el que se midió la fosforilación de sustratos de 136
serín-treonín quinasas (STK) y 126 proteín tirosín-quinasas (PTK). El análisis del quinoma
(figura 5) permitió concluir que en las células L929dt existe una actividad exacerbada de las
PTK y una actividad menor de las STK. Dentro de las PTK se observó una alteración
especialmente elevada en la ruta de señalización del EGFR (figura 6). En esta ruta una de
las proteínas más afectadas es FAK. El nivel de fosforilación de FAK es 8 veces superior en
las células L929dt y es uno de los sustratos que mayor diferencia exhibe.
Página 8Figura 5. Comparación de la fosforilación de sustratos de PTK (A) y de STK (B) entre las
células L929 y las L929dt. El color azul representa que el nivel de fosforilación de un
sustrato es menor y el color rojo que la fosforilación es mayor. (Marco-Brualla y Anel,
comunicación personal).
Figura 6. Esquema de las rutas de señalización relacionadas con el EGFR alteradas en las
células L929dt respecto a las L929. El color azul representa que el nivel de fosforilación de
un determinado sustrato es menor y el color rojo que la fosforilación es mayor para ese
sustrato. Cuanto más lleno está el termómetro más acusada es esa diferencia de
fosforilación entre las líneas celulares. (Marco-Brualla y Anel, comunicación personal).
Página 93. Objetivos
1) Validar la diferencia en actividad PTK observada en el estudio del quinoma en este
modelo celular.
2) Estudiar el efecto de la inhibición general de proteínas tirosín quinasas (PTKs) en este
modelo celular.
3) Validar la hiperfosforilación en Y397 de FAK observada en el estudio del quinoma:
3.1) Cuantificar su expresión en este modelo celular.
3.2) Estudiar el efecto del defactinib (inhibidor de la fosforilación de FAK)
independientemente o combinado con el DCA.
3.3) Estudiar la localización intracelular de FAK en estas células
Enmarcado dentro del proyecto del Ministerio de Ciencia e Innovación “Searching for
efficient antitumoral combinations: metabolic and tyrosine kinase inhibitors, immunogenic
chemotherapy and allogeneic NK cells (CHEMIMMUNODEATH)”.
4. Materiales y métodos
4.1 Cultivos celulares: uso y mantenimiento para experimentos
La línea celular L929dt (del inglés “detached”) se obtuvo a partir de la línea celular
L929 de células de fibroblasto de ratón transformadas12. Los cíbridos dtL929 y L929dt se
obtuvieron a partir de las L929 y las L929dt. Tanto las líneas celulares de fibroblasto de
ratón L929 y L929dt, así como los cíbridos dtL929 y L929dt se cultivaron en una estufa a 37ºC
y 5% de CO2 en medio de cultivo DMEM completo: DMEM GlutaMAX 4’5% D-glucopiranosa
(Gibco, Waltham, MA, USA) suplementado con Suero Fetal Bovino (SFB) (Sigma) al 5% y
un coctel antibiótico compuesto de penicilina/estreptomicina al 1% (Sigma). El manejo de
las líneas celulares se llevó a cabo en una campana de flujo laminar vertical (Telstar) para
mantener la asepsia.
Para evitar que las células alcanzaran un estado de confluencia, se realizaron pases
aproximadamente cada 48 h. Cada vez que se hicieron los pases de las células adherentes
(L929 y dtL929), se incubaron las células con una solución de tripsina/EDTA (PAN Biotech) a
37ºC, se lavaron con PBS y se diluyó 1 ml de células en 7 ml de medio DMEM completo. En
el caso de las células en suspensión (L929dt y L929dt) se diluyeron aproximadamente 1 ml
de células en 10ml de medio DMEM completo.
Página 10La viabilidad de las células se midió con colorante azul Trypan en proporción 1:1
(suspensión de células:solución de Trypan). Este colorante les da a las células una
tonalidad azulada si puede atravesar su membrana y esto solo ocurre si las células no están
vivas. Para realizar el contaje se utilizó una cámara de Neubauer y un microscopio óptico
(Nikon Eclipse 50i).
Los stocks de estas líneas celulares se almacenaron en tanques criopreservadores
de Nitrógeno líquido a -196ºC (Air Liquide) en criotubos (Nunc®, Thermo Scientific).
4.2 Generación de cíbridos transmitocondriales
La generación de cíbridos transmitocondriales dtL929 y L929dt se realizó por nuestro
grupo de investigación22 como se detalla en Marco-Brualla, Joaquín et al. (2019) en
colaboración con el grupo GENOXPHOS. Para generar los cíbridos dtL929 se trataron las
células L929dt (donantes de núcleo) con una dosis letal de rodamina 6 G y se fusionaron
con citoplastos (sin núcleo) provenientes de las células L929. Para generar los cíbridos
L929dt se fusionaron en presencia de polietilenglicol (PEG) las mitocondrias de las células
L929dt con células L929 que habían perdido las mitocondrias.
4.3 Experimentos con inhibidores
4.3.1 Estudio del inhibidor general de PTKs genisteína
Para cada una de las 4 líneas celulares, se sembraron 3 × 104 células por pocillo en
una placa de fondo plano de 96 pocillos; en presencia o ausencia del inhibidor de PTKs
genisteína. El fármaco se utilizó a dosis de 200 nM, 500 nM, 1 µM, 10 µM y 50 µM y se
incubaron durante 24 h a 37ºC.
Para poder evaluar la muerte celular tras incubación con el fármaco, las células se
marcaron simultáneamente con anexina-FITC (Inmunostep) y 7-Aminoactinomycin D
(Biolegend) durante 10 minutos en oscuridad. Finalmente, se analizaron mediante citometría
de flujo (FACSCalibur, BD Bioscience).
4.3.2 Efecto del defactinib en el modelo celular L929
Por cada una de las 4 líneas celulares, se sembraron 3 × 104 células por pocillo en
una placa de fondo plano de 96 pocillos, en presencia o ausencia de defactinib (VS-6063,
PF-04554878), un inhibidor de la fosforilación de FAK en Y39739,40. El fármaco se utilizó a
dosis de 1 µM, 2 µM, 5 µM y 10 µM y se incubó durante 24 h a 37ºC. La muerte celular se
evaluó por citometría de flujo como se ha indicado en el apartado 4.3.1.
Página 114.3.3 Efecto de la terapia combinada: defactinib y DCA
Para el experimento de terapia combinada se sembraron 2 × 104 células en una
placa de 96 pocillos y se incubaron con DCA (5 y 15 mM) durante 48 h. Tras 48 h de
incubación se renovaron las dosis de DCA, se añadió el Defactinib (10 µM) y se incubó
durante 24 h suplementarias. La muerte celular se evaluó por citometría de flujo como se ha
indicado en el apartado 4.3.1.
4.3.4 Ensayos de inhibición del crecimiento mediante la técnica de MTT
El efecto del defactinib en la inhibición del crecimiento se evaluó mediante la técnica
de Mosmann adaptada a microplacas41. El reactivo MTT es reducido en cristales de azul de
formazán principalmente por la actividad mitocondrial y/o lisosomal de las células vivas, lo
que permite evaluar la viabilidad celular. Para ello se sembraron 3 × 104 células por pocillo
en una placa de fondo plano de 96 pocillos y se incubaron con varias dosis de defactinib
(1, 2, 5 y 10 µM) durante 24 h. Pasado el tiempo de incubación, se añadió a cada pocillo 10
µL de una solución MTT (5 mg/ml) y se incubó durante 3 horas suplementarias a 37ºC. A
continuación, los cristales se disolvieron con 100 µl de una mezcla de isopropanol/HCl 0.01
M y se analizó la absorbancia de cada uno de los puntos mediante un espectrofotómetro
(Dynatech, Pina de Ebro, Spain) a una longitud de onda de 550 nm.
4.4 Análisis de expresión de proteínas mediante Western Blot
Se extrajeron las proteínas de aproximadamente 8 × 106 células para cada una de
las cuatro líneas celulares (L929, L929dt, dtL929 y L929dt). En primer lugar, se sembraron
2 × 106 células por línea y se dejaron incubando durante dos días. En segundo lugar, se
centrifugó el contenido de cada frasco de cultivo a 1500 rpm durante 5 minutos y se
resuspendió el pellet celular con PBS. Este proceso (centrifugar y resuspender en PBS) se
repitió varias veces para eliminar el medio de cultivo mediante lavados con PBS. En tercer
lugar, se eliminó el sobrenadante y se añadió tampón de lisis y se dejó en hielo. En cuarto
lugar, pasados 30 minutos se centrifugaron las muestras a 12000 rpm durante 30 minutos.
El sobrenadante recuperado se cuantificó mediante un ensayo BCA (Thermo Fisher,
Rockford, USA). El objetivo del ensayo es cuantificar la proteína total de cada línea celular
mediante la absorbancia que genera el reactivo BCA. Se siguió el protocolo para comparar
la absorbancia que generaron las proteínas de las cuatro líneas celulares dentro de una
recta de calibrado de proteína conocida. Después, se igualó la cantidad de proteína total en
todas las muestras.
Página 12La electroforesis de las muestras, previamente hervidas a 95ºC durante 5-10
minutos y centrifugadas 1 minuto a 1000rpm, se hizo en un gel al 8% de poliacrilamida-SDS
adecuado para una proteína de alto peso molecular. En cada carril (uno por línea celular) se
añadieron 10 µl de proteína. Se emplearon cubetas de la marca Bio-Rad (Life Sciences,
California, USA) y el gel se dejó correr aproximadamente 70-80 minutos a unos 180 V y 20
mA en un tampón de electroforesis.
A continuación se realizó una transferencia semiseca de las proteínas a una
membrana de nitrocelulosa durante 1 hora y 15 minutos, se bloqueó la membrana con una
dilución de tampón B con leche al 5%, y se dejó incubando durante la noche en cámara fría
a 4ºC con el anticuerpo primario anti-FAK (Cell Signaling, #3285), anti-fosfoFAK (Cell
Signaling, #3283) y anti-fosfotirosina (Clone, 4G10, Upstate) Al día siguiente, tras tres
lavados de 5 minutos, se incubó la membrana con el anticuerpo secundario conjugado con
la peroxidasa de rábano (anti IgG de conejo para la proteína FAK y anti IgG de ratón para la
β-actina) a una dilución 1:2000 durante aproximadamente 60 minutos a temperatura
ambiente en el agitador. Después de otros tres lavados se añadió luminol como sustrato de
la peroxidasa (Pierce® ECL Western Blotting Substrate, Thermo Scientific, Rockford, USA)
para poder detectar por quimioluminiscencia el anticuerpo primario y se reveló utilizando el
equipo Amersham Imager 680 (GE Healthcare Life Sciences).
4.5 Estudio de la localización de FAK mediante microscopía de fluorescencia
Para el experimento se sembraron 2 × 105 células en una placa de 24 pocillos sobre
un cubreobjetos redondo, previamente esterilizados con etanol, y se incubaron durante 24 h
para que estas se adhirieran al cubre. Al día siguiente, se centrifugó la placa durante 3
minutos a 3500 rpm y se eliminó el sobrenadante por aspiración. Seguidamente, las células
se incubaron 10 minutos a temperatura ambiente 300 µl de paraformaldehido (PFA) al 4%
para fijarlas. Una vez fijadas las células, se centrifugó de nuevo la placa durante 3 minutos a
3500 rpm, se eliminó el sobrenadante y se lavó el PFA con PBS. A continuación se
permeabilizó con una mezcla de saponina y PBS al 1% que se dejó incubando a
temperatura ambiente durante 20 minutos, se lavó con PBS y se añadió el anticuerpo
primario anti-FAK (Cell Signaling, #3285) a una dilución 1:800 en PBS al 5% SFB, que se
dejó incubando durante una hora. Tras centrifugar de nuevo la placa durante 3 minutos a
3500 rpm, eliminar el sobrenadante y lavar con PBS se añadió una mezcla de PBS con BSA
al 2%, hoescht para teñir los núcleos 10 µg/ml y el anticuerpo secundario IgG de conejo
conjugado con FITC (Invitrogen, A11034) a la dosis de 4 µg/ml y se dejó incubando una
hora en oscuridad, a temperatura ambiente. El excedente de PBS se lavó con agua miliQ.
Página 13Para observar las tinciones en el microscopio de fluorescencia se utilizó el fluoromont G
como amplificador de la señal.
4.6 Análisis estadístico
Se ha utilizado un valor p5.1.2 Inhibición de las PTKs con genisteína
En segundo lugar, utilizando la genisteína, se comprobó que las líneas celulares en
suspensión L929dt y L929dt eran más sensibles a la inhibición de las PTKs. Este resultado
indica que estas células metastásicas son significativamente más dependientes de la
sobreactivación de las PTK observada previamente en el análisis del quinoma.
Figura 8. Efecto citotóxico de la genisteína sobre las 4 líneas celulares L929, L929dt, L929dt
y dtL929 en dosis de 200 nM a 50 µM cuantificado mediante citometría de flujo. Los
experimentos se realizaron un mínimo de 2 veces para cada línea celular (n≥2).
Página 155.2 Niveles de expresión de FAK y FAK fosforilada en Y397
Seguidamente, se procedió a estudiar la expresión de la proteína FAK y de su nivel
de fosforilación en Y397 mediante inmunoblot.
Figura 9. Expresión de FAK y FAK fosforilada en Y397 en las líneas celulares L929, L929dt,
L929dt y dtL929 comparando con la β-actina como control de carga. El Western Blot se realizó
un mínimo de 3 veces para cada línea celular (n=3).
Se puede observar que hay una mayor fosforilación en Y397, así como un mayor
nivel de expresión de FAK en las dos líneas celulares en suspensión L929dt y L929dt (figura
9). Este resultado confirma los resultamos del quinoma y además evidencia un aumento en
la expresión de FAK en las dos líneas L929dt y L929dt. En realidad, parece que las líneas
celulares adherentes L929 y dtL929 apenas expresan esta quinasa.
5.3 Experimentos de citotoxicidad
Se utilizó el inhibidor de FAK defactinib39,40 para cuantificar el efecto citotóxico y
citostático que puede tener la inhibición de la fosforilación de esta quinasa en este modelo
celular.
5.3.1 Muerte celular
Como se puede observar en la figura 10 el defactinib no indujo muerte celular de
forma significativa en ninguna de las líneas ensayadas.
Página 16Figura 10. Efecto del defactinib sobre las 4 líneas celulares L929, L929dt, L929dt y dtL929 en
dosis de 1 a 10 µM cuantificado mediante citometría de flujo. Los experimentos se
realizaron un mínimo de 3 veces para cada línea celular (n=3).
5.3.2 Inhibición del crecimiento
Con el fin de evaluar si el defactinib inhibía el crecimiento, a pesar de que este no
afecte significativamente a la muerte celular, se realizaron ensayos de reducción del MTT
en las 4 líneas celulares L929, L929dt, L929dt y dtL929. En este caso, los resultados
obtenidos muestran que existe un mayor efecto citostático en las líneas celulares con
defectos mitocondriales L929dt y L929dt con respecto a las adherentes L929 y dtL929,
aunque este efecto sólo es significativo a la concentración de 10 µM.
Figura 11. Efecto citostático del defactinib sobre las 4 líneas celulares L929, L929dt, L929dt y
dtL929 en un rango de dosis de 1 a 10 µM cuantificado por espectrofotometría. Los
experimentos se realizaron un mínimo de 3 veces para cada línea celular (n=3).
Página 17Atendiendo a este resultado, el rol de FAK estaría principalmente relacionado con
una contribución al aumento de la capacidad proliferativa en las células L929dt,
probablemente a través de la vía EGFR.
5.3.3 Efecto combinado del defactinib y el DCA.
Figura 12. Efecto del defactinib (Dfk) sobre las 4 líneas celulares L929, L929dt, L929dt y
dtL929 en dosis de 10 µM y del DCA a dosis subletales de 5 mM y 15 mM cuantificado
mediante citometría de flujo. Los experimentos se realizaron un mínimo de 3 veces para
cada línea celular (n=3).
Dados los resultados obtenidos en el apartado 5.4.2 se decidió combinar el
defactinib a la dosis más alta (10 µM), con el inhibidor metabólico DCA a dosis subletales
(5 mM y 15 mM) sobre las 4 líneas celulares. A pesar de demostrar que el defactinib
sensibiliza a las células en suspensión (figura 11), no se observó un efecto sinérgico de la
combinación de DCA y defactinib. En concreto, el efecto de ambos fármacos combinados
fue similar al del DCA en ausencia de defactinib en todas las líneas celulares (figura 12).
5.4 Estudio de la localización de FAK mediante microscopía de fluorescencia
Seguidamente se estudió la localización de FAK en las células L929 y en las células
en suspensión L929dt por microscopía de fluorescencia. Como se muestra en la figura 13,
FAK se localiza en el núcleo de las células L929dt y en cambio en las células L929 se
observa un leve marcaje que no es nuclear.
Página 18Esta observación podría estar asociada tanto al estrés oxidativo que se produce como
consecuencia de los defectos mitocondriales en estas dos líneas celulares22 como a la
pérdida de adherencia, que podría desencadenar la traslocación de FAK al núcleo35.
L929 L929dt
Figura 13. Comparación de la localización celular de FAK en las líneas celulares L929 y
L929dt. En azul, el campo de células con los núcleos marcados con hoescht, y justo debajo
en verde, el mismo campo pero marcado con el anticuerpo anti-FAK y el anticuerpo
secundario conjugado con FITC.
Así mismo, los cíbridos L929dt y dtL929 se reproducen los fenotipos de las líneas
celulares donantes de mitocondrias (figura 14). En las células L929dt se reproduce la
localización nuclear de FAK observada en las L929dt mientras que en las células dtL929 se
observa el tenue marcaje extracelular de FAK observado en las células L929. Por lo que
estos resultados permiten asociar los defectos mitocondriales de las líneas celulares en
suspensión L929dt y L929dt con la traslocación de FAK al núcleo.
Página 19
25 µmdtL929 L929dt
Figura 14. Comparación de la localización celular de FAK en los cíbridos L929dt y dtL929. En
azul, el campo de células con los núcleos marcados con hoescht, y justo debajo en verde, el
mismo campo pero marcado con el anticuerpo anti-FAK y el anticuerpo secundario
conjugado con FITC.
Si bien la traslocación nuclear de FAK observada no tiene que ver con su actividad
quinasa, ya que se ha demostrado que estas células son ligeramente sensibles al
defactinib, podría tratarse de otra actividad tumorigénica en la que se podría estar
promoviendo la degradación de p53 y en consecuencia, se estaría promoviendo
principalmente la resistencia a la apoptosis19, 35.
Página 206. Conclusiones
Con los resultados de este trabajo se validaron algunos de los resultados obtenidos
en el estudio del quinoma en este modelo celular. Se ha confirmado que en la línea celular
L929dt existen tanto mayores niveles de proteínas fosforiladas en tirosina como una mayor
fosforilación y expresión de FAK que en las células parentales L929. Además, se pueden
asociar estos resultados a sus defectos mitocondriales dado que estos se reprodujeron de
forma análoga en el cíbrido L929dt, que lleva las mitocondrias de las células L929dt y el
núcleo de las células parentales L929, pero no en la línea celular dtL929, que lleva las
mitocondrias de las células parentales L929 y el núcleo de las células L929dt.
Por otra parte, se ha demostrado la localización nuclear de FAK en las células
L929dt y L929dt así como que la inhibición de la actividad de FAK a través del fármaco
defactinib no es especialmente efectiva para eliminar estas células, ya sea en solitario o en
combinación con dicloroacetato (DCA). Además, se propone el uso de inhibidores de EGFR
así como inhibidores de otras proteínas asociadas para poder indagar en las bases
moleculares de la conversión espontánea de las células L929 al fenotipo L929dt el cual es
responsable de su mayor proliferación, supervivencia y agresividad tumoral.
6. Conclusions
The results obtained in this project validated some of the data from the quinome
analysis. It was confirmed that L929dt cells had more protein tyrosine kinases fosforilated
and also that FAK expression was higher and more fosforilated in this cells than parental
L929 cells.
Moreover, this results can correlate with the defects in mtDNA because they were
similar on L929dt cells which had L929dt mtDNA but were different on dtL929 cells, which had
L929dt nuclear DNA but L929 mtDNA.
Furthermore, we found that FAK was located in the nucleus in both L929dt and
dt
L929 cells as well as that FAK inhibition was not really effective to eliminate this cells
neither with defactinib nor with the antimetabolic drug dichloroacetate or the combination of
both. Hence, we propose EGFR inhibitors and/or another associated protein inhibitors to
better understand the molecular basis of the spontaneous transition of L929 cells to L929dt,
cells which is reponsible of their high proliferation, survival and tumoral behaviour.
Página 217. Bibliografía
1. Weitzman, J B, and M Yaniv. “Rebuilding the road to cancer.” Nature vol. 400,6743
(1999): 401-2.
2. Fares, Jawad et al. “Molecular principles of metastasis: a hallmark of cancer revisited.”
Signal transduction and targeted therapy vol. 5,1 28. 12 Mar. 2020
3. Hanahan, Douglas, and Robert A Weinberg. “Hallmarks of cancer: the next generation.”
Cell vol. 144,5 (2011): 646-74.
4. Lee, Eva Y H P, and William J Muller. “Oncogenes and tumor suppressor genes.” Cold
Spring Harbor perspectives in biology vol. 2,10 (2010): a003236
5. Singh, Rumani et al. “Regulation of apoptosis in health and disease: the balancing act of
BCL-2 family proteins.” Nature reviews. Molecular cell biology vol. 20,3 (2019): 175-193.
6. Strumane, K et al. “Cadherins in cancer.” Handbook of experimental pharmacology ,165
(2004): 69-103.
7. De Palma, Michele et al. “Microenvironmental regulation of tumour angiogenesis.”
Nature reviews. Cancer vol. 17,8 (2017): 457-474.
8. Rundhaug, Joyce E. “Matrix metalloproteinases and angiogenesis.” Journal of cellular
and molecular medicine vol. 9,2 (2005): 267-85
9. Fagiani, Ernesta, and Gerhard Christofori. “Angiopoietins in angiogenesis.” Cancer
letters vol. 328,1 (2013): 18-26.
10. Sweeney, Melanie D et al. “Pericytes of the neurovascular unit: key functions and
signaling pathways.” Nature neuroscience vol. 19,6 (2016): 771-83.
11. Muenst, S et al. “The immune system and cancer evasion strategies: therapeutic
concepts.” Journal of internal medicine vol. 279,6 (2016): 541-62.
12. Catalán, Elena et al. “MHC-I modulation due to changes in tumor cell metabolism
regulates tumor sensitivity to CTL and NK cells.” Oncoimmunology vol. 4,1 e985924. 3
Feb. 2015
13. Warburg, O. “On the origin of cancer cells.” Science (New York, N.Y.) vol. 123,3191
(1956): 309-14.
14. Koppenol, Willem H et al. “Otto Warburg's contributions to current concepts of cancer
metabolism.” Nature reviews. Cancer vol. 11,5 (2011): 325-37
15. Liberti, Maria V, and Jason W Locasale. “The Warburg Effect: How Does it Benefit
Cancer Cells?.” Trends in biochemical sciences vol. 41,3 (2016): 211-218
16. Tirpe, Alexandru Andrei et al. “Hypoxia: Overview on Hypoxia-Mediated Mechanisms
with a Focus on the Role of HIF Genes.” International journal of molecular sciences vol.
20,24 6140. 5 Dec. 2019
Página 2217. Muñoz-Pinedo, C et al. “Cancer metabolism: current perspectives and future
directions.” Cell death & disease vol. 3,1 e248. 12 Jan. 2012
18. Marbaniang, Casterland, and Lakhan Kma. “Dysregulation of Glucose Metabolism by
Oncogenes and Tumor Suppressors in Cancer Cells.” Asian Pacific journal of cancer
prevention : APJCP vol. 19,9 2377-2390. 26 Sep. 2018,
19. Gottlieb, Eyal, and Karen H Vousden. “p53 regulation of metabolic pathways.” Cold
Spring Harbor perspectives in biology vol. 2,4 (2010): a001040.
20. Blackshear, P J et al. “Treatment of severe lactic acidosis with dichloroacetate.” Diabetes
care vol. 5,4 (1982): 391-4.
21. Stacpoole, Peter W et al. “Controlled clinical trial of dichloroacetate for treatment of
congenital lactic acidosis in children.” Pediatrics vol. 117,5 (2006): 1519-31.
22. Marco-Brualla, Joaquín et al. “Mutations in the ND2 Subunit of Mitochondrial Complex I
Are Sufficient to Confer Increased Tumorigenic and Metastatic Potential to Cancer
Cells.” Cancers vol. 11,7 1027. 21 Jul. 2019
23. Tataranni, Tiziana, and Claudia Piccoli. “Dichloroacetate (DCA) and Cancer: An
Overview towards Clinical Applications.” Oxidative medicine and cellular longevity vol.
2019 8201079. 14 Nov. 2019
24. Sutendra, Gopinath, and Evangelos D Michelakis. “Pyruvate dehydrogenase kinase as a
novel therapeutic target in oncology.” Frontiers in oncology vol. 3 38. 7 Mar. 2013
25. Lemmon, Mark A, and Joseph Schlessinger. “Cell signaling by receptor tyrosine
kinases.” Cell vol. 141,7 (2010): 1117-34
26. Ségaliny, Aude I et al. “Receptor tyrosine kinases: Characterisation, mechanism of
action and therapeutic interests for bone cancers.” Journal of bone oncology vol. 4,1 1-
12. 23 Jan. 2015
27. Sulzmaier, Florian J et al. “FAK in cancer: mechanistic findings and clinical applications.”
Nature reviews. Cancer vol. 14,9 (2014): 598-610.
28. https://www.omim.org/entry/600758 (consultado el 17 de Mayo de 2021)
29. https://www.uniprot.org/uniprot/Q05397 (consultado el 17 de Mayo de 2021)
30. http://www.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG0000016939
8;r=8:140657900-141002216 (consultado el 17 de Mayo de 2021)
31. Fang, Xu-Qian et al. “Somatic mutational analysis of FAK in breast cancer: a novel gain-
of-function mutation due to deletion of exon 33.” Biochemical and biophysical research
communications vol. 443,2 (2014): 363-9.
32. https://www.proteinatlas.org/ENSG00000169398-PTK2/tissue (consultado el 17 de Mayo
de 2021)
Página 2333. Crompton, Brian D et al. “High-throughput tyrosine kinase activity profiling identifies FAK
as a candidate therapeutic target in Ewing sarcoma.” Cancer research vol. 73,9 (2013):
2873-83.
34. Ok Atılgan, Alev et al. “Association between focal adhesion kinase and matrix
metalloproteinase-9 expression in prostate adenocarcinoma and their influence on the
progression of prostatic adenocarcinoma.” Annals of diagnostic pathology vol. 45 (2020):
151480.
35. Lim, Ssang-Taek Steve. “Nuclear FAK: a new mode of gene regulation from cellular
adhesions.” Molecules and cells vol. 36,1 (2013): 1-6.
36. Chen H., Cheng C.Y. Focal Adhesion Kinase (FAK). In: Choi S. (eds) Encyclopedia of
Signaling Molecules. Springer, Cham. (2018)
37. Grigera, Pablo R et al. “FAK phosphorylation sites mapped by mass spectrometry.”
Journal of cell science vol. 118,Pt 21 (2005): 4931-5.
38. Paoli, Paolo et al. “Anoikis molecular pathways and its role in cancer progression.”
Biochimica et biophysica acta vol. 1833,12 (2013): 3481-3498.
39. Zhang, Lingyuan et al. “Focal adhesion kinase (FAK) inhibitor-defactinib suppresses the
malignant progression of human esophageal squamous cell carcinoma (ESCC) cells via
effective blockade of PI3K/AKT axis and downstream molecular network.” Molecular
carcinogenesis vol. 60,2 (2021): 113-124.
40. Gerber, David E et al. “Phase 2 study of the focal adhesion kinase inhibitor defactinib
(VS-6063) in previously treated advanced KRAS mutant non-small cell lung cancer.”
Lung cancer (Amsterdam, Netherlands) vol. 139 (2020): 60-67.
41. Alley, M C et al. “Feasibility of drug screening with panels of human tumor cell lines
using a microculture tetrazolium assay.” Cancer research vol. 48,3 (1988): 589-601
Página 24También puede leer

















































