Enfermedades neurometabólicas: Orientación diagnóstica a partir de la clínica neurológica - fi-admin
←
→
Transcripción del contenido de la página
Si su navegador no muestra la página correctamente, lea el contenido de la página a continuación

Ponencias Pediátr Panamá 2017; 46 (2): 112-118
Enfermedades neurometabólicas García- Méndez
Enfermedades neurometabólicas: Orientación diagnóstica a partir
de la clínica neurológica.
Autores: Dr. Leonardo García- Méndez 1
Dr. María G. Jiménez-Méndez 2
Recibido para publicación: 15 de Mayo 2017
Aceptado para publicación: 21 de Mayo 2017
Resumen
Las Enfermedades Neurometabólicas (ENM) son trastornos bioquímicos en la estructura y/o función de proteínas, de origen genético, por
mutaciones en el ADN, causando un déficit enzimático con acumulación del sustrato o producción de metabolitos anormales. Consideradas
enfermedades raras por su escasa incidencia, en su conjunto constituyen un motivo de admisión, consulta y morbimortalidad infantil frecuente.
Las formas clínicas de presentación incluyen la forma de “intoxicación”, las que afectan el metabolismo energético y las que afectan las moléculas
complejas con compromiso de las organelas celulares (lisosomas, peroxisomas). El diagnóstico preciso y oportuno de las ENM es crucial,
especialmente para aquellos trastornos que son tratables o manejables, con menor impacto en los pacientes, evitando no solo la muerte sino
secuelas irreversibles. El diagnóstico clínico es el primer paso para su abordaje partiendo de la elaboración de una adecuada historia clínica,
recolección detallada de antecedentes familiares y personales, considerando la edad de la aparición de los síntomas, siendo el periodo neonatal
donde la mayoría debutan. La exploración física completa por aparatos y sistemas con especial énfasis en el examen neurológico, permite
solicitar con mayor precisión los exámenes complementarios correspondientes para realizar una aproximación diagnóstica más acertada. El
uso de herramientas diagnósticas mediante programas electrónicos, algunos de ellos disponibles gratuitamente como el Programa
NeurometPlus (www.neurometplus.com), permite alcanzar de forma rápida y efectiva el diagnóstico más probable, así como manejo médico y
tratamiento disponible actual.
Palabras Clave: neurometabólicas, neuropediatría, diagnóstico clínico.
Abstract
Neurometabolic Disorders are conditions caused by alterations in the biochemical structure and function of proteins resulting from changes in
DNA, causing a lack or dysfunction of an enzyme leading to an accumulation of a substrate or production of abnormal metabolites. Though
considered rare diseases, due to low incidence, together they constitute a frequent cause for pediatric admissions and consultations with
significant morbidity and mortality in childhood. Neurometabolic disorders may be classified as disorders of intoxication, energy metabolism, or
those disorders affecting complex molecules compromising the cellular organelles (lysosomes and peroxisomes). A timely and precise diagnosis
is crucial especially in treatable conditions in order to avoid irreversible sequelae and even death. When encountering a child with a potential
neurometabolic disorder, making a clinical diagnosis is the first step which should include an adequate clinical history, taking into consideration
the age of onset of symptoms, and a detailed family history. The neonatal period is when the majority of these conditions make their debut. A
complete physical exam with special emphasis on the neurological exam allows for greater precision to help guide the complementary exams
that correspond to each condition in order to reach the most probable diagnosis. The use of diagnostic tools including electronic programs and
databases, some of which are free of charge such as NeurometPlus (www.neurometplus.com), allow a quick and effective diagnosis, along with
a guide for medical management and current treatment regimens available.
Keywords: neurometabolic, neuropediatrics, clinical diagnosis.
Conflictos de interés: Los autores certifican que no existen conflictos de interés que impidan la correcta publicación de este
artículo y que el artículo es original y no ha sido publicado previamente en ninguna revista científica médica.
1
Neurólogo Pediatra y Medicina de Sueño. Pediatric Neurology Clinic. McAllen, Texas, USA.
2
Neurólogo Pediatra. Investigadora Neuroclínica. Pediatric Neurology Clinic.McAllen ,Texas ,USA.
Correo electrónico:leogarciamendez@gmail.com
112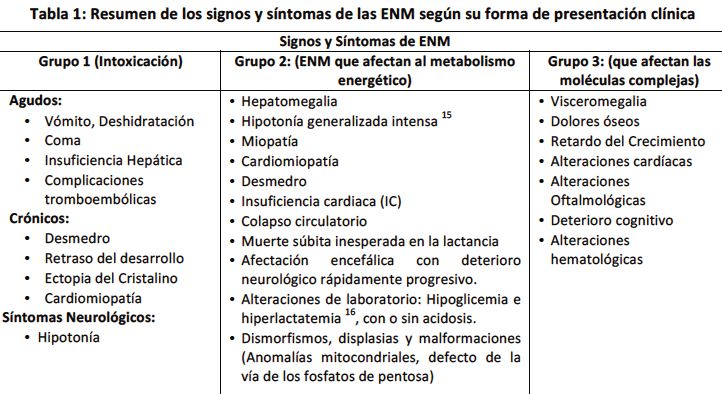
Ponencias Pediátr Panamá 2017; 46 (2): 112-118
Enfermedades neurometabólicas García- Méndez
Introducción pediatra y al neuropediatra una mejor base para el diagnóstico,
A principio del siglo XX Sir Archibald Garrod en su discurso de manejo y tratamiento.11 El diagnóstico preciso y oportuno de las
1908 al Colegio Real de Médicos de Londres, utilizó la expresión ENM es crucial, especialmente para aquellos trastornos que son
Error Congénito del Metabolismo (ECM) para describir un grupo tratables o manejables, con menor impacto en el individuo,
de trastornos (alcaptonuria, pentosuria benigna, albinismo y evitando no solo la muerte sino secuelas irreversibles. 10,12
cistinuria) aparentemente causados por defectos puntuales en el
metabolismo, debido a fallas o ausencias de una enzima que Datos clínicos que apoyan la existencia de una ENM en
cataliza un paso específico en una ruta metabólica 1, señalando niños con problemas neurológicos
que eran enfermedades de por vida y hereditarias.1
Antecedentes Familiares
La secuenciación del genoma humano ha permitido estudiar • Consanguinidad: la existencia de algún grado de
los ECM o Enfermedades Neurometabólicas (ENM) definidas consanguinidad es importante determinar en todo niño son
como trastornos bioquímicos en la estructura y/o función de sospecha de ENM, dado que la mayoría son AR. 2 Realizar el
proteínas, de origen genético, por mutaciones en el ADN. 2 Dicha árbol genealógico es una herramienta útil.
mutación puede causar un déficit enzimático con acumulación • Antecedentes Familiares: interrogar si existen familiares con el
del sustrato, producción de metabolitos anormales o reducción mismo proceso, hermanos en casos de herencia AR, con patrón
del producto. Otras veces, la proteína implicada se encarga del de transmisión vertical en casos de herencia autosómica
transporte transmembrana y origina un déficit de la sustancia dominante (AD), ligada a X (de madres a varones), o mitocondrial
que debería ser transportada. Por último, el cambio estructural de (maternal en algunos casos del ADN mitocondrial)
la proteína puede conllevar un aumento de la actividad • Historia Familiar de abortos, muertes súbitas o niños fallecidos
enzimática y un exceso de producto 3,4. Con mucha frecuencia de causa no aclarada.13
son monogénicas y de herencia autosómica recesiva (AR). 2
Antecedentes Personales
Desde un punto de vista fisiopatológico, los ECM pueden • Período libre de síntomas
dividirse en defectos en la síntesis o catabolismo de moléculas • Sepsis neonatal “sin etiología clara”
complejas, defectos en metabolismo intermedio y • Relación de síntomas con cambios en la dieta
deficiencias en la producción o utilización de energía.5 En • Rechazo a la alimentación
consecuencia, pueden involucrar múltiples órganos y • Alteraciones bioquímicas características (acidosis,
sistemas, siendo las manifestaciones neurológicas y las hiperamonemia, Hipoglicemia, etc.)
digestivas las más frecuentes. 4 • Episodios neurológicos agudos
• Regresión de funciones neurológicas
Hoy en día se calcula que existen más de 700 ECM, con más de • Falla de Crecimiento no relacionado con malnutrición
600 genes conocidos 6, y el número va en aumento constante • Historia de afectación de órganos, aparatos y sistemas.
debido a la identificación de nuevos trastornos metabólicos. 6, 7 • Dismorfias faciales (DF)
Aunque considerados individualmente los ECM son
enfermedades raras, en su conjunto constituyen más de 5% de Formas de Presentación Clínica de ENM
las admisiones en un hospital pediátrico, con una fre¬cuencia en Desde una perspectiva fisiopatológica, las ENM se dividen en
la población general de cerca de 1/2000 nacidos vivos 8, de ellos tres grupos 5:
50% desarrolla la enfermedad durante el período neonatal, de los Grupo 1: trastornos que dan lugar a “intoxicación”. Abarca ECM
cuales solamente una quinta parte tienen tratamiento más o intermediario que conducen a intoxicación aguda o progresiva
menos efectivo 9, siendo su incidencia difícil de precisar debido a por acumulación de sustancias tóxicas proximales al bloqueo
los escasos datos estadísticos.10 metabólico. Se incluyen
a. Las ECM del catabolismo de los Aminoácidos (AA):
Desde la perspectiva clínica, la aproximación diagnóstica en fenilcetonuria (PKU) incluida en el screaning neonatal 14,
neuropediatría debe plantearse mediante una adecuada enfermedad de la orina con olor de jarabe de arce (EOJA),
secuencia diagnóstica, donde la sospecha clínica, tomando en homocistinuria, tirosinemia, etc.
cuenta la edad del paciente, los hallazgos clínicos, permitan b. La mayoría de las acidurias orgánicas, galactosemia,
solicitar los exámenes complementarios necesarios ofreciendo al intolerancia hereditaria a la fructosa.
113
Ponencias Pediátr Panamá 2017; 46 (2): 112-118
Enfermedades neurometabólicas García- Méndez
c. Las intoxicaciones por metales ( Wilson, Menkes, 1. Síntomas tempranos o antenatales: se destacan: a)
hemocromatosis) y porfirias. Malformaciones verdaderas: malformaciones esqueléticas,
cardiopatías congénitas, aplasias viscerales y defectos del
Todos comparten semejanzas clínicas: no interfieren con el tubo neural, b) Displasias: corticales, quistes corticales,
desarrollo embrionario-fetal y se presentan con un intervalo anomalías de fosa posterior, riñones poliquísticos, quistes
libre de síntomas y con signos clínicos de “intoxicación” (Ver hepáticos y c) Signos funcionales: Retardo del Crecimiento
tabla 1). Aunque la fisiopatología es algo diferente, en este intrauterino (RCIU), hidropesía fetal, hepatoesplenomegalia,
grupo también se incluyen los errores congénitos de la microcefalia.17
síntesis y catabolismo de los neurotrasmisores (monoaminas, 2. Crisis agudas y recurrentes: comienzo más tardío de los
GABA y Glicina), errores de la síntesis de aminoácidos (serina, síntomas: coma, ataxia, vómitos, acidosis. Intolerancia al
glutamina y prolina/ornitina). ejercicio, IC, falla renal, hepática u otras vísceras.
Grupo 2: trastornos que afectan al metabolismo 3. Síntomas neurológicos crónicos y progresivos: muchas
energético. Los síntomas se deben a una deficiencia de la veces inadvertidos por años como algunos síntomas
producción o utilización de la energía dentro del hígado, gastrointestinales, 18 y otros síntomas más llamativos como:
miocardio, músculo, encéfalo u otros tejidos (Ver tabla 1). hipotonía, demora del desarrollo, trastorno del desarrollo
Producen enfermedad multisistémica y miopatía. 10 Se intelectual, epilepsia, deterioro neurológico, signos
dividen a su vez en defectos de la energía mitocondrial más psiquiátricos.
graves y en general no tratables y de la energía citoplásmica
menos graves e incluyen: trastornos de la glucólisis, del 4. Presentaciones específicas y permanentes de órganos y
metabolismo del glucógeno y la glucogénesis, sistemas: cardiológicas, dermatológicas, endocrinológicas,
hiperinsulinismo, anomalías del metabolismo de la creatina. gastrointestinales, hematológicas, etc. 2.
Grupo 3: trastornos que afectan la síntesis o catabolismo Signos clínicos específicos: cuando se enuncia que se debe
de moléculas complejas, afectando las organelas celulares. tener un buen olfato para el diagnóstico de las ENM, es una
Los síntomas son permanentes, progresivos, independientes expresión literal debido a que muchas cursan con un olor
de acontecimientos intercurrentes y no relacionados con la particular o especial tanto corporal como de la orina (Ver
ingesta de alimentos. Pertenecen a este grupo todos los tabla 2), por lo que es otro aspecto clínico importante para
trastornos con almacenamiento lisosómico, anomalías el abordaje diagnóstico y la descripción de la ENM.
peroximales, desórdenes del tráfico y el procesamiento
intracelulares, como la deficiencia de la α1 antitripsina, Aproximación clínica según síntomas sospechosos por
trastornos congénitos de la glucosilación y los errores aparatos y sistemas
congénitos de la síntesis de colesterol. 2 (Ver tabla 1) Las ENM, pueden cursar de forma multisistémica afectando
múltiples órganos de forma simultánea,
otras sin embargo tienen preferencia
por determinado órgano donde de
forma preferencial son las principales
manifestaciones. Las manifestaciones
neurológicas son muy floridas. A
continuación se resumen los principales
síntomas por aparatos y sistemas de
ENM más comunes.
Circunstancias Clínicas: las ENM se presentan en cuatro grupos de circunstancias clínicas, que deben
ser tomadas en cuenta ante la sospecha diagnóstica: 114Ponencias Pediátr Panamá 2017; 46 (2): 112-118
Enfermedades neurometabólicas García- Méndez
manifestaciones absolutamente inespecíficas a síntomas
cardíacos, respiratorios, hepáticos, digestivos, renales o
hematológicos, con presentación multisistémica. 19
Se debe sospechar una ENM ante todo deterioro
neurológico rápido y progresivo sin causa aparente (en
ausencia de infección, de pérdida del bienestar fetal, de
distocia del parto o de prematuridad). De los grupos clínicos
antes expuestos (Grupo I, II y III), un 70% de los recién nacidos
(RN) inicia los primeros síntomas en forma de “Intoxicación”. Se
trata en general de RN aparentemente sanos, que tras un
intervalo libre de pocas horas a varios días, inician una clínica
de deterioro neurológico progresivo, lento al inicio, con
rechazo de alimento, vómitos, fallo de medro y muy
especialmente alteraciones del tono muscular con manifiesta
y casi constante hipotonía. Rápidamente se instaura un grave
deterioro neurológico (convulsiones, depresión del sensorio y
coma), con una “catástrofe” metabólica (acidosis metabólica,
cetosis, hipoglucemia, aumento del ácido láctico,
hiperamonemia, trombocitopenia, coagulopatía) y evolución
fatal si no se sospecha la enfermedad y se inicia un
tratamiento precoz. El intervalo entre el nacimiento y los
síntomas clínicos pueden oscilar entre horas y semanas,
dependiendo de la naturaleza del bloqueo metabólico y del
medio ambiente. 2
Un 15% manifiesta un alarmante déficit energético. 20 Se
manifiesta entonces un fallo agudo hepático, miocárdico,
muscular, renal o del sistema nervioso. Lógicamente, las
manifestaciones pueden ser tan variadas como retardo,
estancamiento ponderal, alteraciones del ritmo respiratorio,
convulsiones, hipotonía, miopatía, cardiomiopatía, fallo
cardíaco o renal, arritmias, defectos de conducción, colapso
circulatorio, muerte súbita, DF e incluso malformaciones que
pueden detectarse prenatalmente por ultrasonografía. En
estas circunstancias suelen existir manifestaciones
bioquímicas más o menos constantes como hipoglucemia
severa, acidosis metabólica y aumento del ácido láctico. La
presentación clínica es con frecuencia menos evocadora y
exhibe una gravedad variable. 2
Y finalmente un 15 % un grupo de miscelánea, con
manifestaciones clínicas muy variadas y que
mayoritariamente comprometen el metabolismo de las
moléculas complejas 20.
Diagnóstico Clínico por Grupos Etarios En este grupo las manifestaciones son aún más variadas
Presentación Neonatal y Lactancia Temprana: e inespecíficas como por ejemplo cataratas, hidrops fetal
Las formas de presentación son muy variadas y van desde (HF), anemia, ictericia colestática, ascitis, edema, trastornos
115Ponencias Pediátr Panamá 2017; 46 (2): 112-118
Enfermedades neurometabólicas García- Méndez
de coagulación, hepatoesplenomegalia o Pérez J y colaboradores analizaron las características
manifestaciones neurológicas como un retardo del clínicas de pacientes adultos con ECM en España con una
desarrollo, micro/macrocefalia, trastornos de la migración revisión de 500 casos 7, siendo los trastornos de los
neuronal o DF. 21 Aminoácidos los más frecuentes (22%), 52% con síntomas
del grupo 1 “intoxicación”. Algunos estudios confirman
Las convulsiones en el período neonatal como forma de demora en el diagnóstico, confirmando que hasta un 53%
expresión de una disfunción neurológica son una de las por ciento de los pacientes son diagnosticados en la vida
manifestaciones neurológicas más comunes en los ENM. adulta. 22 En un paciente adulto con encefalopatía o coma
Algunos pacientes debutan con crisis en los primeros días y inexplicable o síntomas neuropsiquiátricos agudos
en general éstas se van instaurando junto a otras obligan a considerar ENM como los trastornos del ciclo de
manifestaciones sistémicas. Predominan las crisis la urea, defectos de la remetilación de la homocisteína,
generalizadas, seguidas de las mioclonías y finalmente del porfirias agudas entre otras.
estado de mal convulsivo neonatal. 19
Examen Físico
Presentación Infantil Después de haber realizado una detallada anamnesis y
A partir de los dos primeros años de vida las manifestaciones revisión de todos los antecedentes, el examen físico
neurológicas de ENM son: retraso psicomotor, hipotonía, completo y enfocado es la clave para llegar a un
regresión neurológica, convulsiones, síntomas neurológicos diagnóstico de manera oportuna. La inspección del estado
deficitarios transitorios, afección cerebelosa, piramidal o general del paciente, su reactividad al medio, estado del
extrapiramidal, leucodistrofias o dismielinizaciones, neuropatías sensorio permite sugerir algún cuadro de intoxicación. 23
y miopatías. El Síndrome de Reye o similar a éste, síndrome de Del mismo modo la presencia de características
muerte súbita del lactante o episodios con riesgo vital 20. Los dismórficas sugieren trastornos de la macromolécula
síntomas digestivos (vómitos, disfunción hepática, fallo de (enfermedad peroxisomal y lisosómicas), enfermedades
medro, ictericia, etc.) son frecuentes4. Algunos síntomas mitocondriales. El examen ocular y el fondo de ojo es la
aparecen en relación con el tipo de alimentación, como la ventana para diagnosticar ENM. Las cataratas en un
intolerancia hereditaria a la fructosa 21 y la galactosemia. neonato es altamente sugestivo de Galactosemia. Tomar
en cuenta el olor anormal del paciente que como ya se
Presentación infancia tardía/adolescencia: Los cuadros mencionó es una característica muy común de los ENM
neurológicos que se instauran en este período suelen ser (ver tabla 2). Valorar la piel si presenta o no ictericia permite
regresivos.5 La aparición de crisis convulsivas agudas, encontrar síntomas que pueden orientar el diagnostico.
inexplicables, recurrentes o refractarias a cualquier edad Valorar sus signos vitales, la taquipnea asociada a acidosis
también debe ser una alarma para ENM. Algunas respiratoria en un neonato enfermo puede sugerir defecto
manifestaciones pueden ser precipitadas por un acontecimiento en el ciclo de la urea. En el examen clínico por sistemas la
intercurrente o sin causa franca. La ingesta excesiva de proteínas, exploración cardiaca en descarte de cardiomegalia,
el ejercicio agotador y condiciones que potencien el cardiomiopatía dilatada puede aumentar la sospecha de
catabolismo de proteínas pueden originar descompensaciones. defecto en la oxidación de ácidos grasos, AG tipo 2, entre
La encefalopatía aguda, la ataxia aguda intermitente o conducta otras. La palpación abdominal en busca de
alterada pueden ser signos de ENM, así como síntomas visceromegalias (hepato o hepatoesplenomegalia) que
psiquiátricos, intolerancia al ejercicio, mioglobinuria recurrente, sugieran enfermedad de depósito lisosomal, galactosemia,
crisis de dolor abdominal recurrente, crisis de vómitos con tirosinemia o trastorno mitocondrial. Finalmente la
letargia, dolor óseo, Síndrome de Reye. Síntomas neurológicos exploración neurológica es fundamental. Evaluar
crónicos y progresivos como deterioro cognitivo, discapacidad cognición, comportamiento, presencia de movimientos
intelectual, demora del desarrollo pueden ser comunes a anormales, características de la marcha, tono, reflejos,
muchas condiciones neurológicas pero obligan a descartar ENM coordinación, la regresión de habilidades ya adquiridas, y si
entre otras. presenta convulsiones es muy importante detallar la
semiología de las crisis e inicio de las mismas. Habrá que
Presentación adulta: La prevalencia de ENM en la población sospechar un ECM cuando se observa compromiso de más
adulta es desconocida en muchos países. de un órgano o sistema. 24
116Ponencias Pediátr Panamá 2017; 46 (2): 112-118
Enfermedades neurometabólicas García- Méndez
Herramientas para la aproximación diagnóstica: Referencias
NeurometPlus 1. Garrod EA. The incidence of alkaptonuria: a study clinical
NeurometPlus es un programa disponible en la web, individually. Yale J Biol Med. 2002; 75(4): 221-231.
gratuito que consta de 630 condiciones clínicas, con 2. Sanjurjo P, Baldellou A. Diagnóstico y tratamiento de las
posibilidad de actualización y de expansión. Una vez enfermedades metabólicas hereditarias 4a ed, Ergon, Madrid,
registrado, el programa puede ser usado de varias maneras. 2014.
Primero, como una colección de condiciones, donde se busca 3. Bearn AG, Miller ED. Archibald Garrod and the development of
el nombre de la patología y puede imprimirse la información, the concept of inborn errors of metabolism. Bull Hist Med.
o bien puede realizarse la búsqueda de palabras o textos 1979;53(3):315-28.
específicos presentes en las condiciones, mediante el uso de 4. Palacios A, García O, García-Silva MT. Diagnóstico de los errores
“operadores” que facilitan dicha búsqueda. Por último, se innatos del metabolismo An Pediatr Contin. 2008;6(6):347-52
puede utilizar el sistema de filtros, seleccionando campos 5. Saudubray J. Clinical approach to inherited metabolic diseases.
específicos presentes en las 3 sub-tablas del programa, las In: Inborn Metabolic Diseases. Diagnosis and Treatment.
cuales son: a) edad de comienzo, b) síntomas y signos, y c) Fernandes J, Saudubray J, Van den Berghe G (eds). 5th edition.
exámenes de laboratorio y otros exámenes especiales Springer-Verlag Berlin, 2012. pp 1-52.
anormales presentes en las condiciones del programa. Una 6. Reid ES, Papandreou A, Drury S et al. Advantages and pitfalls of
vez seleccionados los campos de búsqueda de cada an extended gene panel for investigating complex
sub-tabla, se activa el botón de búsqueda y el programa neurometabolic phenotypes. Brain 2016;139(11): 2844–2854.
mostrará solo las condiciones que contienen los campos 7. Pérez-López J, Ceberio-Hualde L, García-Morillo JS et al. Clinical
solicitados, haciendo una lista del diagnóstico diferencial characteristics of adult patients with inborn errors of
solicitado. Contiene así mismo imágenes, videos que facilitan metabolism in Spain: A review of 500 cases from university
la aproximación diagnóstica. Una forma sencilla y rápida tan hospitals. Mol Genet Metab Rep. 2017; 10:92–95
solo con proporcionar los datos de un paciente, y le brindará 8. Hoffman GF. Selective screening for inborn errors of metabolism:
la información de los exámenes complementarios a solicitar y past, present and future. Eur J Pediatr .1994; 153(7 Suppl 1): S2-8.
si dispone o no de tratamiento médico o dietético para su 9. Rebage V, López J, Baldellou A. Enfermedades metabólicas de
abordaje. 11 presentación neonatal, en Enfermedades metabólicas
hereditarias en Sanjurjo P, Baldellou A. Ed Ergon, 2014
Conclusiones: 10. Campistol J. Orientación diagnóstica de las enfermedades
Las ENM son enfermedades raras, en su conjunto neurometabólicas basada en la clínica, estudios metabólicos y
constituyen un motivo de admisión, consulta y neuroimagenológicos. Medicina. 2013; 73(Supl. I):55-62.
morbimortalidad infantil. El diagnóstico preciso y 11. García-Méndez L. NeurometPlus: enfoque práctico del manejo
oportuno de las ENM es crucial, especialmente para de niños con enfermedades neurometabólicas y
aquellos trastornos que son tratables o manejables, con neurogenéticas. Rev Chil Pediat.2016; 87(1):16-19.
menor impacto en el individuo, evitando no solo la muerte 12. Németh AH, Kwasniewska AC, Lise S et al. Next generation
sino secuelas irreversibles 11,12. Este diagnóstico parte de la sequencing for molecular diagnosis of neurological disorders
elaboración de una adecuada historia clínica, con la using ataxias as a model. Brain 2013; 136 (Pt 10): 3106–18.
recolección detallada de los antecedentes familiares y 13. van Sprosen FJ, Molendijk H, Erwich JJ, Smit GP. Inherited
personales, detallar la edad de la aparición de los síntomas, metabolic disease and pregnancy: consequences for mother
ya que la clasificación por grupos etarios permite descartar and child. Ned Tijdschr Geneeskd. 2003;147(6):235-40.
un gran número de ellas, la exploración física completa por 14. Therrell BL, Padilla CD, Loeber JG et al. Current status of
aparatos y sistemas con especial énfasis en el examen newborn screening worldwide: 2015. Semin Perinatol. 2015;
neurológico donde mayormente presenta sus 39(3):171-187.
manifestaciones. El uso de herramientas diagnósticas 15. Moggio M, Colombo I, Peverelli L et al. Mitochondrial disease
como el Programa NeurometPlus disponible de forma heterogeneity: a prognostic challenge. Acta Myol. 2014;
gratuita por internet: www.neurometplus.com, permite 33(2):86-93.
alcanzar el diagnóstico más probable, así como su manejo 16. Ogier de Baulny H. Management and emergency treatment of
médico y posibilidad de tratamiento de forma rápida y neonates with suspicious of inborn errors of metabolism.
efectiva. Semin Neonatol 2002; 7(1): 17-26.
117Ponencias Pediátr Panamá 2017; 46 (2): 112-118
Enfermedades neurometabólicas García- Méndez
17. Cabello JF, Giugliani R. Errores innatos del Metabolismo. Rev.
Med. Clin. Condes - 2015; 26(4) 483-486.
18. Saudubray JM, Nassogne MC, de Lonlay P, Touati G. Clinical
approach to inherited metabolic disorders in neonates: an
overview. Semin Neonatol 2002; 7(1): 3-15.
19. Campistol J. Enfermedades metabólicas en el periodo neonatal
con presentación neurológica. Medicina. 2007; 67 (6/1):
561-568.
20. Leonard JV, Morris AA. Diagnosis and early management of
inborn errors of metabolism presenting around the time of
birth. Acta Paediatr. 2006;95(1):6-14.
21. Tran C. Inborn Errors of Fructose Metabolism. What Can We
Learn from Them? Nutrients 2017; 9 (4): pii: E356. doi:
10.3390/nu9040356.
22. Sirrs S, Hollak C, Merkel M et al. The frequencies of different
inborn errors of metabolism in adult metabolic centres: report
from the SSIEM Adult Metabolic Physicians Group. JIMD Rep.
2016;27:85-91. doi: 10.1007/8904_2015_435. Epub 2015 Oct 9.
23. Mohamed S. Recognition and diagnostic approach to acute
metabolic disorders in the neonatal period. Sudan J Paediatr
2011; 11(1): 20-28.
24. Bravo P, Castro G. Actualización en el manejo agudo de errores
congénitos del metabolismo. Rev Chil Pediatr 2014; 85 (4):
421-427.
118También puede leer

















































