SEGUIMIENTO CARDIOLÓGICO DE LA DISTROFIA MUSCULAR DE DUCHENNE - RECOMENDACIONES DE LA SOCIEDAD ESPAÑOLA DE CARDIOLOGÍA PEDIÁTRICA Y CARDIOPATÍAS ...
←
→
Transcripción del contenido de la página
Si su navegador no muestra la página correctamente, lea el contenido de la página a continuación
SEGUIMIENTO CARDIOLÓGICO
DE LA DISTROFIA MUSCULAR
DE DUCHENNE
RECOMENDACIONES DE LA SOCIEDAD ESPAÑOLA
DE CARDIOLOGÍA PEDIÁTRICA Y CARDIOPATÍAS
CONGÉNITAS
SEGUIMIENTO CARDIOLÓGICO
DE LA DISTROFIA MUSCULAR
DE DUCHENNE
RECOMENDACIONES DE LA SOCIEDAD ESPAÑOLA
DE CARDIOLOGÍA PEDIÁTRICA Y CARDIOPATÍAS
CONGÉNITAS
1.ª edición: febrero de 2021 © de los autores Maquetación e impresión: Lúa Ediciones 3.0, S.L. ISBN: 978-84-121659-7-5 Depósito legal: M-1992-2021 Las ciencias de la salud están en permanente cambio y los autores han verificado que toda la informa- ción se ajuste a los estándares en el momento de la publicación, no obstante antes de su utilización en pacientes debe contrastarse con las recomendaciones actuales. Todos los derechos reservados. Queda prohibida la reproducción total o parcial de esta obra. Ni la totalidad, ni parte de este libro pueden reproducirse o transmitirse por ningún tipo de procedimiento electrónico y mecánico, incluidos los de fotocopia, grabación magnética o cualquier almacenamiento de información y sistema de recuperación, sin permiso previo y escrito de Lúa Ediciones 3.0, S.L.
Autores
Javier Pérez-Lescure Picarzo: Cardiología Infantil, Hospital Universitario Fundación Alcorcón,
Madrid. Profesor de la Facultad de Medicina, Universidad Rey Juan Carlos. Secretario de
la Sociedad Española de Cardiología Pediátrica y Cardiopatías Congénitas. Grupo de Car-
diología Clínica de la Sociedad Española Cardiología Pediátrica y Cardiopatías Congénitas
María Teresa Fernández Soria: Cardiología Infantil, Hospital Universitario Fundación Alcor-
cón, Madrid. Profesora de la Facultad de Medicina, Universidad Rey Juan Carlos. Grupo
de Cardiología Clínica de la Sociedad Española Cardiología Pediátrica y Cardiopatías
Congénitas.
Ana Patricia Fariña Ruiz: Cardiología Infantil y Cardiopatías Congénitas, Hospital Universita-
rio Nuestra Señora de Candelaria, Santa Cruz de Tenerife. Grupo de Cardiología Clínica de
la Sociedad Española de Cardiología Pediátrica y Cardiopatías Congénitas.
Carlos Labrandero de Lera: Cardiología Infantil, Hospital Universitario La Paz, Madrid. Grupo
de Insuficiencia Cardiaca de la Sociedad Española de Cardiología Pediátrica y Cardiopa-
tías Congénitas.
Sergi César Díaz: Cardiología Infantil, Hospital Sant Joan de Déu, Barcelona. Universitat de
Barcelona. Acreditación europea en Ecocardiografía de las cardiopatías congénitas. Pos-
grado en Genética Cardiovascular. Diploma de competencia en Diseño y Análisis de In-
vestigaciones Clínicas.
Israel Valverde: Jefe del Servicio de Cardiología Infantil, Hospital Virgen del Rocío, Sevilla.
Profesor de Pediatría, Universidad de Sevilla-IBIS. Honorary Senior Lecturer-Biomedical
Engineering & Imaging Sciences, King’s College, Londres, Reino Unido. Grupo de Imagen
de la Sociedad Española de Cardiología Pediátrica y Cardiopatías Congénitas.
Carlos Alcalde Martín: Cardiología Infantil, Servicio de Pediatría, Hospital Universitario Río
Hortega, Valladolid. Grupo de Cardiología Clínica de la Sociedad Española Cardiología
Pediátrica y Cardiopatías Congénitas.
Begoña Manso García: Cardiología Infantil, Hospital Universitario Virgen del Rocío, Sevilla.
Junta Directiva de la Sociedad Española Cardiología Pediátrica y Cardiopatías Congé-
nitas. Grupo de Cardiología Clínica e Imagen de la Sociedad Española de Cardiología
Pediátrica y Cardiopatías Congénitas.
David Crespo Marcos: Cardiología Infantil, Hospital Universitario Fundación Alcorcón, Ma-
drid. Profesor de la Facultad Medicina, Universidad Rey Juan Carlos. Grupo de Cardio-
logía Clínica de la Sociedad Española Cardiología Pediátrica y Cardiopatías Congénitas.
Miguel Ángel Martínez Granero: Neurología Infantil, Hospital Universitario Fundación Alcor-
cón, Madrid. Profesor de la Facultad Medicina, Universidad Rey Juan Carlos.
5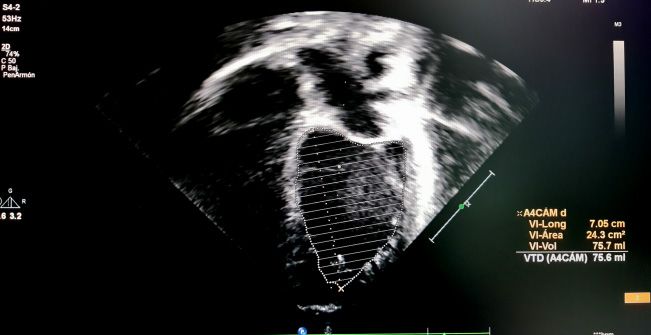
Siglas y acrónimos
AI: aurícula izquierda.
ARA: antagonistas del receptor de la angiotensina.
ARM: antagonista del receptor de los mineralocorticoides.
CK: creatina-quinasa.
DAI: desfibrilador automático implantable.
DAV: dispositivos de asistencia ventricular.
DDVI: diámetro diastólico del ventrículo izquierdo.
DMD: distrofia muscular de Duchenne.
DSVI: diámetro sistólico del ventrículo izquierdo.
IC: insuficiencia cardiaca.
IECA: inhibidores de la enzima convertidora de la angiotensina.
MPI: Miocardial Performance Index o índice de Tei.
Onda A: velocidad del llenado ventricular activo (por contracción auricular) medida por
Doppler pulsado.
Onda E: velocidad del llenado ventricular pasivo medido por Doppler pulsado.
Onda a’: velocidad del movimiento diastólico tardío del anillo mitral medido por Doppler
tisular.
Onda e’: velocidad del movimiento diastólico precoz del anillo mitral medido por Doppler
tisular.
Onda S’: velocidad del movimiento sistólico por Doppler pulsado tisular en el anillo mitral.
RMC: resonancia magnética cardiaca.
SECPCC: Sociedad Española de Cardiología Pediátrica y Cardiopatías Congénitas.
TCI: tiempo de contracción isovolumétrica.
TRI: tiempo de relajación isovolumétrica.
VI: ventrículo izquierdo.
VNI: ventilación no invasiva.
7
Prólogo
Cuando se diagnostica a un niño o una niña una distrofia muscular de Duchenne se ge-
neran muchas incertidumbres, no solo en la familia, sino en los profesionales sanitarios que
tendrán que cuidarles. Una de esas incógnitas es la posibilidad de desarrollar alteraciones
cardiovasculares, que pueden llegar a ser, junto con las respiratorias, las causas de una mala
evolución.
La guía con recomendaciones que ha elaborado la Sociedad Española de Cardiología
Pediátrica y Cardiopatías Congénitas (SECPCC), encabezada por el Dr. Javier Pérez-Lescure,
junto a un numeroso grupo de expertos de la SECPCC, tiene como fin aportar evidencia
científica y estandarizar el manejo de las complicaciones cardiológicas en estos pacientes.
Quiero agradecerles a todos ellos el esfuerzo de revisión y sistematización que han hecho en
esta puesta al día práctica.
Creo que es muy relevante que todos los profesionales conozcamos mejor la enfermedad
y su afectación cardiovascular, que se apliquen los mismos protocolos y tratamientos actuali-
zados. También que exista una coordinación interdisciplinar con otros especialistas, o en red,
con otros centros. Esta guía servirá, no solo para aumentar el conocimiento actual de las dis-
trofinopatías entre los cardiólogos pediatras, sino que espero que estimule la investigación
en este campo. Tenemos retos actuales y futuros que van desde la genética a la medicina
personalizada en la búsqueda de tratamientos específicos.
El objetivo final de esta guía de seguimiento cardiológico no es otro que mejorar la cali-
dad de vida y el pronóstico de pacientes pediátricos y adolescentes con distrofia muscular
de Duchenne, así como disminuir las incertidumbres de ellos y sus familiares. Espero y deseo,
en mi nombre y el de la sociedad que represento, que sirva para avanzar en esa dirección.
Agradezco a las familias de los pacientes su labor diaria, su experiencia es fundamental
en lo que todos pretendemos que es cuidar lo mejor posible y ofrecer la mejor calidad de
vida a los niños y niñas con distrofia muscular de Duchenne.
Por último, quiero también agradecer al laboratorio PTC Therapeutics Spain su colabora-
ción en la edición y difusión de esta guía.
Madrid, 15 de enero de 2021
Dr. Constancio Medrano López
Presidente de la Sociedad Española
de Cardiología Pediátrica y Cardiopatías Congénitas
9Índice
1. Distrofia muscular de Duchenne ........................................................................................................................... 13
1.1. Diagnóstico de la DMD .................................................................................................................................................................. 13
1.2. Evolución natural de la DMD ................................................................................................................................................. 15
1.3. Tratamiento no cardiológico ................................................................................................................................................. 16
1.3.1. Corticoides ....................................................................................................................................................................................... 16
1.4. Nuevos tratamientos ....................................................................................................................................................................... 17
2. Afectación cardiológica ............................................................................................................................................... 19
2.1. Evaluación cardiológica ............................................................................................................................................................... 20
2.1.1. Exploración física .................................................................................................................................................................... 20
2.1.2. Presión arterial .......................................................................................................................................................................... 21
2.1.3. Electrocardiograma ............................................................................................................................................................ 21
2.2. Calendario de revisiones ........................................................................................................................................................... 21
2.2.1. Evaluación inicial .................................................................................................................................................................... 21
2.2.2. Seguimiento reglado ....................................................................................................................................................... 21
2.2.3. Valoración prequirúrgica ........................................................................................................................................... 22
2.3. Mujeres portadoras de la mutación de la distrofina ............................................................................. 22
2.3.1. Afectación de músculo esquelético en mujeres portadoras de la mutación
de la distrofina .......................................................................................................................................................................................... 22
2.3.2. Afectación cardiológica en mujeres portadoras de la mutación
de la distrofina .......................................................................................................................................................................................... 23
2.3.3. Recomendaciones de seguimiento en mujeres portadoras de la mutación
de la distrofina .......................................................................................................................................................................................... 24
3. Evaluación de la función cardiaca por imagen ...................................................................................... 25
3.1. Valoración de la función por ecocardiografía ................................................................................................ 25
3.1.1. Valoración de la función sistólica del ventrículo izquierdo ............................................. 25
3.1.2. Valoración de la función diastólica del ventrículo izquierdo ....................................... 30
3.1.3. Valoración funcional del ventrículo derecho .................................................................................... 34
3.2. Diagnóstico por imagen de la asincronía ventricular en la DMD ........................................ 35
3.3. Resonancia magnética cardiaca ..................................................................................................................................... 36
3.4. Técnicas de imagen con estrés miocárdico ..................................................................................................... 37
3.5. Resumen de las pruebas diagnósticas por imagen en paciente con DMD ............ 37
4. Tratamiento cardiológico ........................................................................................................................................... 39
4.1. Introducción ............................................................................................................................................................................................... 39
4.2. Medicamentos ......................................................................................................................................................................................... 39
11Seguimiento cardiológico de la distrofia muscular de Duchenne
4.2.1. Diuréticos ......................................................................................................................................................................................... 39
4.2.2. Inhibidores del sistema renina-angiotensina-aldosterona (SRAA) ................... 40
4.2.3. Betabloqueantes .................................................................................................................................................................. 41
4.2.4. Digoxina ........................................................................................................................................................................................... 43
4.2.5. Antiarrítmicos ........................................................................................................................................................................... 43
4.2.6. Anticoagulación .................................................................................................................................................................... 43
4.2.7. Medicamentos inotrópicos ...................................................................................................................................... 43
4.2.8. Nuevos fármacos para el tratamiento de la insuficiencia cardiaca (IC) ..... 44
4.3. Intervenciones no farmacológicas para la IC refractaria ............................................................... 44
4.3.1. Ventilación no invasiva (VNI) ................................................................................................................................ 44
4.3.2. Desfibrilador automático implantable (DAI) y resincronización
cardiaca .............................................................................................................................................................................................................. 45
4.3.3. Dispositivos de asistencia ventricular (DAV) ................................................................................. 45
4.3.4. Cuidados paliativos .......................................................................................................................................................... 49
4.3.5. Trasplante cardiaco .......................................................................................................................................................... 50
4.3.6. Tratamiento inotrópico parenteral domiciliario .......................................................................... 51
4.4. Resumen de las recomendaciones ............................................................................................................................. 51
4.5. Otros tratamientos ........................................................................................................................................................................... 54
4.5.1. Corticoides ..................................................................................................................................................................................... 54
4.5.2. Tratamiento respiratorio ............................................................................................................................................ 55
5. Anexo .............................................................................................................................................................................................. 57
6. Bibliografía ............................................................................................................................................................................... 65
12Distrofia muscular de Duchenne
1. Distrofia muscular de Duchenne
Las distrofinopatías son un grupo de El objetivo de este documento es
desórdenes genéticos caracterizados por abordar la afectación cardiaca de la DMD
la aparición de debilidad muscular progre- y emitir las recomendaciones de la Socie-
siva en relación con degeneración de la fi- dad Española de Cardiología Pediátrica y
bra muscular. Cardiopatías Congénitas (SECPCC) en re-
lación a la prevención, diagnóstico precoz
Están causadas por la ausencia, dismi- y tratamiento de las complicaciones car-
nución o expresión anómala de la distrofi- diovasculares como guía para los profesio-
na, una proteína de la membrana citoplas- nales sanitarios en la toma de decisiones
mática de las células musculares, esencial en los aspectos relevantes que conciernen
para la estabilidad estructural del músculo. al manejo de la enfermedad.
La distrofina está codificada por un gen
(gen DMD) localizado en el cromosoma X
(Xp21). Los defectos genéticos que afec- 1.1. Diagnóstico de la DMD
tan al gen DMD pueden ser deleciones
(65%), duplicaciones (5-10%) y mutacio- El diagnóstico precoz juega un papel
nes puntuales (20%)1. decisivo en el manejo de los pacientes con
DMD.
La variante clínica más frecuente es la
distrofia muscular de Duchenne (DMD), La sospecha clínica generalmente co-
con una prevalencia de aproximadamente mienza en la primera infancia, a partir de
5 por cada 100 000 varones y una inciden- los 2-3 años, con alteraciones a nivel del
cia de 1 por cada 3800-6300 recién naci- desarrollo motor. Se debe sospechar una
dos varones2. distrofinopatía en cualquiera de las si-
guientes situaciones4:
Clínicamente, se caracteriza por una
debilidad muscular progresiva de inicio en 1. C
ualquier evidencia de retraso mo-
la infancia, con aparición posterior de com- tor en un niño con historia familiar de
plicaciones músculo-esqueléticas, respira- DMD.
torias y cardiacas que ocasionan discapaci-
dad, dependencia y mortalidad temprana3. 2.
Retraso motor como retraso en el
inicio de la deambulación hasta los
La afectación cardiaca es una de las 16-18 meses, dificultad para levantar-
principales causas de morbimortalidad en se del suelo, correr o saltar, pseudo-
estos pacientes. En términos generales, se hipertrofia de gemelos en ausencia
caracteriza por una disfunción ventricu- de historia familiar.
lar izquierda progresiva que evoluciona a
miocardiopatía dilatada con el desarrollo
de insuficiencia cardiaca y arritmias.
13Seguimiento cardiológico de la distrofia muscular de Duchenne
3.
Aumento inexplicable de transami- transaminasas (GOT, GPT) que, de forma
nasas. errónea, pueden conducir a pensar en una
enfermedad hepática en pacientes presin-
En la Tabla 1 se describen los signos y tomáticos, retrasando el diagnóstico1.
síntomas más frecuentes en la DMD4.
El diagnóstico de certeza se obtiene,
Ante la sospecha clínica, debe solicitarse en la mayoría de los casos, mediante el es-
la medición de niveles de creatina-quinasa tudio genético, evitando la biopsia muscu-
(CK) en sangre. Los niveles de CK se elevan lar, que ha quedado en un segundo plano;
antes del inicio de los síntomas, incluso en es útil en los casos con estudio genético
el periodo neonatal, aumentando progresi- negativo y alta sospecha clínica4.
vamente, con valores 10-100 veces por en-
cima del límite de la normalidad, alcanzado Dado que, aproximadamente un 70-
un pico hacia los 2 años de edad y posterior- 75% de los pacientes con DMD presentan
mente van descendiendo con la edad hasta deleciones o duplicaciones en el gen de la
normalizarse a medida que el tejido muscu- distrofina, el test genético que se usa de
lar ha ido siendo sustituido por tejido fibro- primer nivel es el Multiplex Ligation-de-
so y grasa1,4. Niveles de CK normales irían en pendent Probe Amplification (MLPA)4. Si
contra del diagnóstico de distrofinopatía y este fuese negativo, se solicitaría test de
obligarían a sospechar otras causas5. secuenciación del gen de la distrofina para
buscar mutaciones puntuales o deleciones
Como consecuencia de la destrucción o duplicaciones más pequeñas, presentes
muscular, se produce también elevación de en 20-30% de los pacientes5.
Tabla 1. Signos y síntomas más frecuentes de la DMD
Clínica motora Clínica no motora
•T rastornos de la marcha •R
etraso cognitivo
•P seudohipertrofia de gemelos •E
stancamiento o retraso ponderal
•S igno de Gowers •T
rastornos de aprendizaje y atención
•C aminar de puntillas •R
etraso en el habla o dificultades de
•R etraso motor grueso articulación
•D ebilidad en el control de la cabeza
•D isminución de la resistencia al ejercicio
• Incapacidad para saltar, correr o trepar
•D ificultad para subir escaleras
•P ie plano
•C aídas frecuentes o torpeza
• Incapacidad para seguir a sus compañeros
•P érdida de habilidades motoras
•D olor muscular o calambres
14Distrofia muscular de Duchenne
1.2. Evolución natural de la DMD una fase de meseta entre los 4-8
años. En este punto se produce
La historia clásica de los pacientes con un deterioro progresivo de las ca-
DMD se puede definir en 3 estadios, mar- pacidades ya adquiridas, con una
cados por la pérdida de la deambulación4: pérdida de la fuerza muscular. Se
manifiestan claramente los sig-
1. Fase presintomática (0-2 años). No nos clásicos de la DMD: signo de
hay manifestaciones clínicas de debi- Gowers positivo, pseudohipertro-
lidad muscular, pero puede aparecer fia muscular y una marcha cada
un retraso en el neurodesarrollo: re- vez más dificultosa. Podemos ob-
traso motor global, retraso en el ha- servar contracturas musculares,
bla, dificultades para la relación-co- fracturas (secundarias a caídas y al
municación. tratamiento con corticoides) y pri-
meros síntomas de escoliosis. Un
2. Fase ambulatoria (2-12 años): 20-34% de los pacientes presentan
alteraciones cognitivo-conductua-
2.1.
Fase ambulatoria temprana (2-5 les: déficit intelectual o síntomas
años). La debilidad muscular afec- de espectro autista.
ta inicialmente con mayor intensi-
dad a las extremidades inferiores, 3. Fase no ambulatoria (12-20 años):
especialmente a la musculatura
proximal. En esta primera etapa 3.1.
Fase no ambulatoria temprana
aparecen los primeros síntomas de (12-16 años): viene definida por la
debilidad muscular: caídas frecuen- pérdida de la marcha, en la mayo-
tes, dificultades para la marcha y ría de los casos ocurre a los 12-14
para subir escaleras. Ya se puede años. Todavía hay capacidad de
apreciar a la exploración la pseudo- mantener la postura. En esta eta-
hipertrofia de gemelos, la debilidad pa se desarrolla la escoliosis como
axial y el signo de Gowers positivo consecuencia de la progresión de
(desde la posición de sentado el la debilidad hacia las extremidades
niño afecto tiene que apoyar las superiores y la afectación de los
manos para erguirse). Es en esta músculos paravertebrales. A su vez,
etapa cuando también empiezan a también surgen las complicaciones
detectarse problemas cognitivos. cardiacas y respiratorias.
2.2.
Fase ambulatoria tardía (5-12 3.2.
Fase no ambulatoria tardía (20
años): en este periodo, los pacien- años): fase de incapacidad para
tes van adquiriendo las habilidades mantenerse sentado. Agravamien-
motoras de forma progresiva, a un to de las complicaciones respira-
ritmo más lento, hasta alcanzar torias, cardiacas y ortopédicas.
15Seguimiento cardiológico de la distrofia muscular de Duchenne
La supervivencia más allá de la recientes recomiendan su continuidad,
tercera década es poco frecuente ya que se ha demostrado que preserva la
debido al fracaso respiratorio o la fuerza en extremidades superiores, redu-
afectación cardiológica. ce la progresión de la escoliosis y retrasa
el desarrollo de problemas respiratorios y
Existe una variabilidad individual en cardiológicos6,7.
el ritmo de progresión y gravedad de los
síntomas influida por factores genéticos y El efecto beneficioso está bien defini-
ambientales1. do, pero existe controversia sobre que cor-
ticoide es el más indicado y en qué dosis.
Los más utilizados son prednisona y defla-
1.3. Tratamiento no cardiológico zacort, y los regímenes más utilizados se
recogen en la Tabla 2.
Actualmente no existe un tratamiento
curativo para la DMD. El pilar fundamental
del manejo de estos pacientes es el trata- Tabla 2. Tipo y dosificación de corticoides
miento con corticoides y el abordaje mul- en la DMD
tidisciplinar de los síntomas y complicacio-
1. P
rednisona, 0,75 mg/kg/día
nes de la enfermedad, que abarca medidas
2. D
eflazacort, 0,9 mg/kg/día
a nivel respiratorio, cardiaco, rehabilitador,
ortopédico, gastrointestinal y nutricional.
1.3.1. Corticoides El tratamiento crónico con corticoides
conlleva una serie de efectos secundarios
Los corticoides constituyen el trata- que presentarán una alta proporción de
miento de base de la DMD. Varios estudios pacientes. Ambos fármacos presentan un
muestran que el uso de este grupo farma- perfil de seguridad similar. Hay estudios
cológico ralentiza la progresión de la en- que afirman que el aumento de peso y la
fermedad, es beneficioso para mejorar la apariencia cushingoide se da con mayor
función motora y pulmonar, la fuerza, re- frecuencia con prednisona que con de-
ducir el riesgo de escoliosis y retrasar la flazacort, mientras que el riesgo de cata-
pérdida de la deambulación6. ratas es mayor con este último7. Se debe
contemplar la posibilidad de los efectos
Se recomienda iniciar el tratamiento secundarios para anticiparlos y prevenirlos
con corticoides cuando la función motriz o disminuirlos cuando sea posible. Si apa-
alcanza un nivel estable (4-6 años), inclu- recen y son inmanejables o intolerables, se
so antes de que se presente un deterioro deberá disminuir la dosis en un 25-30%. Si
físico sustancial6. fuese necesario se podría cambiar a otro
Existe controversia sobre su uso tras régimen de dosificación antes de valorar
la pérdida de la deambulación; estudios su retirada completa.
16Distrofia muscular de Duchenne
1.4. Nuevos tratamientos de la distrofina, en pacientes ambu-
lantes a partir de 2 o más años de
Se están desarrollando nuevas tera- edad. El eteplirsen ha permitido recu-
pias farmacológicas que buscan modificar perar la pauta de lectura en algunas
la historia de la enfermedad, actuando de deleciones específicas de la región
dos formas diferentes8: central del gen en algunos casos de
DMD.
•T
erapias modificadoras del gen DMD,
cuya finalidad es la recuperación de •T
erapias modificadoras de las con-
la formación de distrofina funcionan- secuencias de la ausencia de distro-
te. El atalureno y el eteplirsen son, de fina: incluyen fármacos dirigidos a
momento, los únicos productos apro- la miostatina, moléculas antiinflama-
bados por las agencias reguladoras torias y antioxidantes, compuestos
europea (Agencia Europea del Me- para reducir la fibrosis, fármacos para
dicamento) y norteamericana (Food mejorar la vasodilatación, mejorar
and Drug Administration), respecti- la función mitocondrial o regular la
vamente. El atalureno está indicado utrofina. Sin embargo, no hay ningún
para el tratamiento de la DMD debida fármaco aprobado actualmente, to-
a una mutación sin sentido en el gen dos en están fase de investigación.
17Afectación cardiológica
2. Afectación cardiológica
Tras la mejora de los cuidados respirato- de satisfacer las necesidades fisiológicas
rios en las últimas décadas, la enfermedad y comienzan a aparecer datos clínicos de
cardiaca es actualmente la principal causa insuficiencia cardiaca (IC). Asimismo, este
de mortalidad de los pacientes con DMD. La miocardio enfermo puede ser el origen de
deficiencia de la distrofina se manifiesta a arritmias potencialmente letales.
nivel cardiaco en forma de miocardiopatía.
Estudios observacionales sugieren que
La distrofina se localiza en la parte inter- el grado de disfunción ventricular en la
na del sarcolema esquelético y cardiaco, y DMD se correlaciona con la edad y la gra-
juega un papel importante en la regulación vedad de la afectación musculoesquelética.
de la transducción de señales. La falta de De esta forma, se describen tres etapas11:
distrofina causa inestabilidad del sarcolema
durante los ciclos de contracción y relaja- • Etapa preclínica: comienzan aparecer
ción, junto con la reducción de la transmi- alteraciones a nivel microscópico, que
sión de fuerza generada por los sarcóme- se manifiestan con alteraciones leves
ros. Además, en el corazón, la ausencia de en el electrocardiograma, disfunción
distrofina también favorece el aumento de diastólica ligera e incluso anomalías
los niveles de calcio intracelular, activando de la movilidad segmentaria del ven-
mecanismos que favorecen la degradación trículo izquierdo. Afecta al 61,5% de
de proteínas contráctiles, promoviendo la los pacientes antes de los 10 años.
muerte celular y la fibrosis9.
• Etapa clínica: progresan las altera-
En análisis anatomopatológicos reali- ciones observando atrofia de car-
zados en muestras de pacientes con DMD, diomiocitos, fibrosis subendocárdica
se ha observado hipertrofia y atrofia de y dilatación de cavidades en el eco-
cardiomiocitos con fibrosis. La fibrosis del cardiograma, a pesar de lo cual to-
ventrículo izquierdo se ha observado tam- davía no se evidencian claramente
bién en autopsias y en estudios con reso- los síntomas en la mayor parte de los
nancia magnética (mediante realce tardío pacientes, prácticamente todos los
con gadolinio) de pacientes con DMD o en pacientes por encima de los 18 años.
mujeres portadoras de la mutación para la
distrofina10. • Etapa de afectación cardiológica evi-
dente: afecta al 57% de los pacientes
Los pacientes con DMD progresiva- por encima de los 18 años en la que
mente desarrollan disfunción ventricular, aparecen síntomas claros.
que se acompaña de dilatación ventricular
en fases avanzadas. A medida que la enfer- A consecuencia de la debilidad muscu-
medad evoluciona, el miocardio es incapaz lar que caracteriza a la DMD, los signos y
19Seguimiento cardiológico de la distrofia muscular de Duchenne
síntomas de IC suelen ser sutiles y difíciles tico (proBNP) y, opcionalmente, la capaci-
de reconocer. Esta circunstancia ha favo- dad funcional basal mediante el cálculo del
recido que los pacientes con DMD clásica- consumo máximo de oxígeno con prueba
mente hayan sido derivados al cardiólogo de esfuerzo cardiopulmonar si están en
de forma tardía, con lo que ello conlleva etapa preclínica y ambulatoria.
para el pronóstico. Por tanto, es importan-
te llevar a cabo una estrategia proactiva ya La ecocardiografía ha sido la prueba de
que el diagnóstico y tratamiento tempra- imagen utilizada clásicamente, pero es habi-
nos son esenciales para optimizar la dura- tual que los pacientes tengan malas venta-
ción y la calidad de vida de los pacientes. nas acústicas, por las deformidades toráci-
Sería ideal la implicación de un cardiólogo cas o la obesidad. En cambio, las imágenes
integrado en un equipo multidisciplinar en obtenidas por resonancia magnética car-
torno a la DMD y con experiencia clínica en diaca (RMC) no dependen de la morfología
el manejo de la IC y de las miocardiopatías del tórax y, además, esta técnica aporta más
asociadas con enfermedades neuromus- información y ha demostrado mayor preci-
culares. Las manifestaciones clínicas espe- sión en la valoración de la función ventricu-
rables se muestran en la Tabla 3. lar en la DMD12. Por todo ello, la RMC es la
prueba de imagen de elección actualmente
en el seguimiento de esta enfermedad. Hay
Tabla 3. Síntomas de insuficiencia cardiaca situaciones en las que se seguirá optando
en los pacientes con DMD por la ecocardiografía como en los pacien-
• Fatiga tes menores de 6-7 años que no suelen ser
• Pérdida de peso capaces de colaborar en la realización del
• Náuseas y vómitos estudio, en caso de intolerancia por parte
• Dolor abdominal del paciente (ya sea postural o psicológica)
• Alteraciones del sueño y ante la no disponibilidad de la técnica.
• Incapacidad para tolerar actividad física
diaria Estos pacientes pueden presentar alte-
• Pueden aparecer síntomas relacionados raciones del ritmo que precisen tratamien-
con alteraciones del ritmo como to específico. Así, el seguimiento debe
palpitaciones, presíncope o síncope incluir la realización de Holter-ECG de 24
horas e incluso pueden ser necesarios mo-
nitores de eventos en algunos casos.
2.1. Evaluación cardiológica
2.1.1. Exploración física
La valoración basal debe incluir historia
médica completa, historia familiar, explo- La exploración puede proporcionar pis-
ración física, electrocardiograma y estudio tas iniciales de la presencia y extensión de
de imagen no invasivo, analítica basal con la enfermedad cardiaca. Los signos vitales
marcadores cardiacos y péptido natriuré- a menudo incluyen taquicardia sinusal en
20Afectación cardiológica
reposo. El examen del cuello puede mostrar onda R alta en V1-V3, ondas Q prominen-
distensión venosa yugular. En la palpación tes en derivaciones laterales (I, aVL, V6),
torácica puede apreciarse un desplaza- inferiores (II, III, aVF) y, menos frecuente-
miento del punto de máximo impulso infe- mente, en derivaciones anteriores (V1-V4)
rior y lateral debido a un ventrículo izquier- así como alargamiento del intervalo QT
do aumentado de tamaño. En la fase aguda corregido. Son menos frecuentes los tras-
de la IC, puede aparecer un ritmo de galope tornos del sistema de conducción, espe-
con tercer ruido. Puede aparecer un soplo cialmente a nivel intraauricular y del nodo
mesosistólico grado I-II/VI en el segundo auriculoventricular (AV) y las arritmias
espacio intercostal izquierdo y un aumen- supraventriculares. Las arritmias ventricu-
to del componente pulmonar del segundo lares graves pueden aparecer en las fases
ruido. La auscultación pulmonar puede avanzadas de la enfermedad en relación
presentar disminución de la ventilación en con disfunción ventricular.
las bases. La hepatomegalia, en caso de es-
tar presente, suele ser difícil de palpar por
la escoliosis. En casos de IC, puede haber 2.2. Calendario de revisiones
edema en los miembros inferiores.
2.2.1. Evaluación inicial
2.1.2. Presión arterial
Todo paciente con diagnóstico confir-
El consumo crónico de corticoides se mado de DMD debe ser remitido para va-
relaciona con el desarrollo de hipertensión loración a la consulta de Cardiología, para
arterial (HTA), a pesar de lo cual, parece que realización de historia cardiológica, historia
los pacientes con DMD tienen tendencia a familiar, exploración, electrocardiograma y
presentar cifras de presión arterial global- prueba de imagen no invasiva (ecocardio-
mente bajas en reposo. Se desconocen los grama en menores de 6-7 años y RMC en
mecanismos que lo justifican, pero podría mayores de dicha edad o en pacientes con
ser multifactorial, por disfunción autonómi- limitación de la ventana acústica), analítica
ca junto con la debilidad muscular que afec- basal con proBNP y, opcionalmente, capa-
tarían a la precarga, la limitación de la movi- cidad funcional basal mediante el cálculo
lidad, la tendencia a ingerir menor cantidad del consumo máximo de oxígeno con prue-
de líquidos y la toma de medicamentos para ba de esfuerzo cardiopulmonar si está en
la IC, que disminuyen la presión arterial13. etapa preclínica y ambulatoria.
2.1.3. Electrocardiograma 2.2.2. Seguimiento reglado
La mayor parte de los pacientes tie- Se revisará al paciente al menos una vez
nen alteraciones en el electrocardiograma al año en la fase ambulatoria y fase ambu-
(ECG). Las más habituales son taquicar- latoria temprana, realizando historia clínica
dia sinusal, acortamiento del intervalo PR, cardiológica, exploración, electrocardiogra-
21Seguimiento cardiológico de la distrofia muscular de Duchenne
ma, prueba de imagen no invasiva y analíti- nal cardiaca mediante eco-RMC de estrés
ca. En la fase ambulatoria tardía y en caso farmacológico en fase no ambulatoria o
de detectar signos o síntomas sugestivos ecografía de estrés con ejercicio físico en
de afectación cardiológica (inicio de sínto- etapa ambulatoria.
mas de IC o alteración en las pruebas de
imagen, como fibrosis miocárdica o dilata-
ción o disfunción del ventrículo izquierdo), 2.3. Mujeres portadoras
se aumentará la frecuencia de las revisio- de la mutación de la distrofina
nes. Con la aparición de las alteraciones
cardiológicas aumenta el riesgo de arrit- Las mujeres familiares de un varón con
mias, por lo que se debe realizar registro diagnóstico de DMD deberían ser analiza-
Holter-ECG de 24 horas en las revisiones a das para detectar el gen. Una vez identifica-
partir de entonces, especialmente en caso das las mujeres portadoras, hay diferentes
de presentar realce tardío en la RMC o dis- opciones reproductivas que, según el deseo
función sistólica ventricular izquierda. En de gestación, deben plantearse: diagnósti-
los pacientes con corticoides, debe descar- co genético preimplantacional y genética
tarse la presencia de HTA. La periodicidad de biopsia coriónica o líquido amniótico.
de realización de la RMC debe establecerse
de forma individualizada, en función de la Las mujeres portadoras de una muta-
clínica, el grado de disfunción, la calidad de ción patogénica tienen riesgo de enferme-
ventana acústica ecocardiográfica, los ha- dad muscular esquelética y miocardiopa-
llazgos ecográficos o de la RMC previa, la tía, aunque la presentación clínica suele
disponibilidad del centro, la necesidad de ser más leve que en los varones y de apa-
sedación o la disponibilidad, entre otros. rición más tardía, a partir de los 25 años14.
La ecografía debe realizarse en todas las
revisiones, incluso en las revisiones en las Se estima que alrededor de un 8% de
que se realice RMC, pues es útil como com- las mujeres portadoras presenta algún
paración de los hallazgos de la RMC para síntoma, como algún grado de debilidad
sucesivas revisiones. muscular con o sin miocardiopatía, aun-
que podría ser mayor, dada la falta de es-
2.2.3. Valoración prequirúrgica tudio y seguimiento en esta población de
portadoras15.
En caso de precisar cirugía mayor,
como cirugía de escoliosis, se deberá rea- 2.3.1. Afectación de músculo esquelético
lizar una valoración previa con electrocar- en mujeres portadoras de la mutación de
diograma y prueba de imagen no invasi- la distrofina
va, ya que el anestesista debe conocer
el riesgo específico del paciente de cara Una forma de presentación descrita en
al procedimiento al que se vaya a some- torno al 17% de las portadoras es la debi-
ter, valorando prueba de reserva funcio- lidad muscular asimétrica de predominio
22Afectación cardiológica
proximal (glúteos, músculos aductores), periores. Al sentarse y levantarse del suelo,
con una edad de debut variable16. El mús- pueden precisar ayuda de los miembros
culo esquelético puede mostrar un patrón superiores14.
de fibras musculares en mosaico, con ni-
veles reducidos de expresión de la distro- 2.3.2. Afectación cardiológica
fina debido a la inactivación sesgada del en mujeres portadoras de la mutación
cromosoma X con un patrón miopático/ de la distrofina
distrófico menos prominente y con una
regeneración focal menor que en varones. En mujeres portadoras se ha detectado
Además, en las biopsias musculares de afectación miocárdica, cuya gravedad po-
mujeres portadoras, puede verse inflama- dría estar relacionada directa y proporcio-
ción del endomisio y perimisio e incremen- nalmente con la edad, aunque otras series
to de la expresión de genes relacionados no encuentran esta relación19,20. A pesar de
con moléculas responsables de fibrosis. no presentar síntomas cardiacos, hasta un
Todo esto sugiere que la fibrosis ocurre 44-60% de las mujeres portadoras podrían
durante la progresión de la enfermedad, manifestar alguna alteración cardiológica,
suele ser de grado leve y sin la presencia como disfunción ventricular.
de necrosis, miofagocitosis ni sustitución
grasa en las biopsias musculares, presente En estudios realizados en mujeres
en varones con DMD17. portadoras, se han identificado alteracio-
nes en la RMC en el 47% de las mujeres
Entre los síntomas más frecuentes es- con disfunción ventricular asintomática y
tán la debilidad y dolor muscular, y calam- fibrosis miocárdica22. Además de los bio-
bres musculares. Hay estudios que sugie- marcadores clásicos como la CK, existen
ren una afectación motora y funcional con otros biomarcadores que aún están en es-
la edad y que, a mayor precocidad en el tudio, como el micro RNA miR-29c, para el
debut de los síntomas, la gravedad de la seguimiento y detección precoz de muje-
manifestación clínica será mayor18. res portadoras23.
Además, también pueden presentar al- Ante la evidencia reciente al respecto,
gunos movimientos compensatorios, como se propone que se haga un seguimiento
ayuda de los miembros superiores y am- a largo plazo, tal y como se realiza en el
pliación de la base de soporte al sentarse o caso de otras miocardiopatías heredita-
levantarse de la silla o del suelo. Al caminar, rias24. A pesar de las recomendaciones
pueden presentar flexión plantar de la ar- que se han estado llevando a cabo, tam-
ticulación del tobillo, hiperextensión de la bién apoyadas por la American Academy
rodilla e inclinación pélvica. Al subir y bajar of Pediatrics, se ha calculado en algunas
escaleras, pueden presentar también incli- series que tan solo del 35-45% de las
nación pélvica y de tronco, hiperextensión portadoras tenía un seguimiento cardio-
de la rodilla y ayuda de los miembros su- lógico, aunque en un reciente estudio se
23Seguimiento cardiológico de la distrofia muscular de Duchenne
eleva hasta el 71%25. Algunos de los mo- 2.3.3. Recomendaciones de seguimiento
tivos detectados por los que no reciben en mujeres portadoras de la mutación
una atención cardiológica son la falta de de la distrofina11,24
conocimiento del riesgo cardiológico, en-
contrarse asintomáticas y para no preo- 1. E
valuación basal cardiovascular en
cupar al resto de la familia. edad adulta joven que incluya:
1.1. ECG de 12 derivaciones.
No existe un consenso específico de 1.2. Imagen cardiaca no invasiva: eco-
tratamiento farmacológico en mujeres cardiografía o, preferiblemente
portadoras en caso de existir miocardio- RMC con contraste para el realce
patía, pero la tendencia es a utilizar inhi- tardío de gadolinio.
bidores de la enzima convertidora de la 1.3. B iomarcadores, según contexto
angiotensina (IECA) o betabloqueantes en clínico.
caso de IC sintomática y siguiendo las di- 1.4. H olter cardiaco de 24 h, según
rectrices de las guías clínicas de IC26. contexto clínico.
2. S eguimiento cardiológico propues-
to: cada 3-5 años.
24Evaluación de la función cardiaca por imagen
3. Evaluación de la función cardiaca por imagen
La realización de pruebas de imagen La RMC es la prueba de referencia en la
capaces de detectar afectación miocárdi- valoración en los pacientes con DMD, pero
ca subclínica y el inicio del tratamiento de su mayor coste, menor disponibilidad y la
forma precoz es fundamental para mejorar necesidad de sedación en pacientes no co-
el pronóstico de los pacientes con DMD, laboradores hacen que, en muchos casos,
que depende cada vez más de la afecta- no pueda realizarse de forma habitual. La
ción cardiológica26. RMC detecta las alteraciones regionales
del ventrículo izquierdo de forma más pre-
Clásicamente, la ecocardiografía ha coz, y aporta otros datos, como la caracte-
sido la técnica habitual de valoración car- rización del miocardio y la función del ven-
diológica, por su accesibilidad, inocuidad trículo derecho. Las secuencias en modo
y bajo coste. A pesar de sus ventajas, tiene cine permiten cuantificar la función y di-
limitaciones importantes que dificultan la mensiones cardiacas, y la administración
práctica diaria, como una menor sensibi- de contraste permite mediante el realce
lidad para detectar cambios mínimos en tardío de gadolinio identificar zonas de
el miocardio, y limitaciones propias del fibrosis, marcador de daño tisular precoz,
paciente con DMD, como la mala venta- muchas veces previo a la afectación de la
na ecográfica, la movilidad reducida, la función ventricular28. Por lo que, siempre
intolerancia al decúbito, la escoliosis o la que esté disponible y valorando los ries-
obesidad, que dificultan la realización de la gos asociados a la necesidad de sedación
prueba en condiciones óptimas. en algunos pacientes, debe ser la prueba
de elección en la mayoría de los casos.
No obstante, la aplicación de técnicas
de mayor resolución y complejidad en la 3.1. Valoración de la función
ecocardiografía convencional 2D y Do- por ecocardiografía
ppler color, como la imagen de Doppler
tisular (IDT) o la medición del strain por 3.1.1. Valoración de la función sistólica
la técnica del speckle tracking, permiten del ventrículo izquierdo.
valorar de forma más adecuada la función
sistólica y diastólica y detectar precoz- La función sistólica del ventrículo iz-
mente la afectación miocárdica. quierdo se puede evaluar mediante modo
M, eco bidimensional y Doppler.
Hay otras opciones invasivas para me-
jorar el rendimiento, como la ecocardio- A. Valoración de la función sistólica por
grafía o la RMC con estrés miocárdico es- cambios dimensionales
timulando el miocardio con dobutamina o
ejercicio, limitado por la capacidad física Se debe determinar la fracción de
de los pacientes. eyección (FE) y la fracción de acortamien-
25Seguimiento cardiológico de la distrofia muscular de Duchenne
to (FA) del ventrículo izquierdo. La FE re- trículo izquierdo en base a los diámetros,
presenta la reducción porcentual del volu- asumiendo que la geometría del ventrículo
men de la cavidad del ventrículo izquierdo es la normal, es decir, una elipse elongada,
durante la sístole. La FA representa la re- por lo que su validez es limitada en caso
ducción porcentual del diámetro del ven- de alteración de la geometría ventricu-
trículo izquierdo durante la sístole. lar. Para su obtención, se deben medir en
modo M los diámetros telesistólico y tele-
LA FE se puede calcular por dos mé- diastólico en eje largo o corto paraesternal
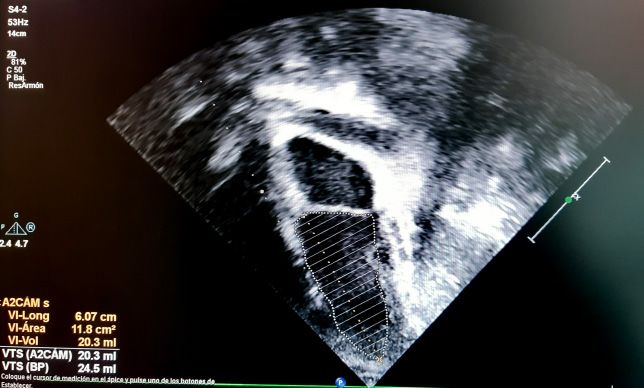
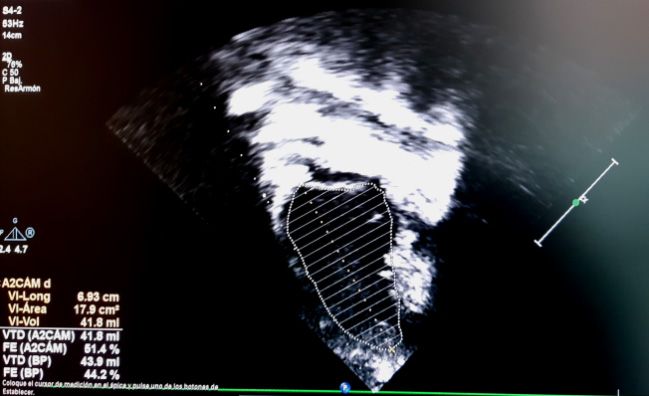
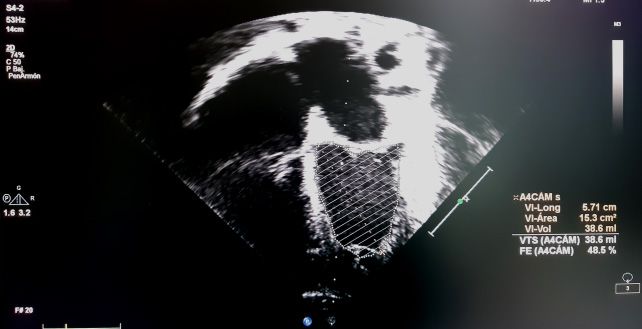
todos, Simpson y Teicholz. En el método (Figura 2). Los valores normales de la FE
de Simpson o método biplano, se debe se sitúan en 56-78%.
medir el área ventricular en el plano api-
cal de 4 cámaras y en el de 2 cámaras La FA se calcula de la misma manera
en sístole y en diástole. Está valoración que la FE por Teicholz, y es la más utilizada
puede ser difícil de realizar en caso de no en Pediatría por su facilidad de obtención,
identificar los bordes de la pared ventri- pero no sería válida en casos de alteración
cular con nitidez, y consume más tiempo de la geometría ventricular o alteraciones
(Figura 1). segmentarias, alteración de la movilidad
del septo, además no refleja el acorta-
El método de Teicholz aplica una fór- miento longitudinal y es dependiente de la
mula que estima los volúmenes del ven- precarga.
Figura 1. Cálculo de la FE por método Simpson en plano de 4 cámaras (imágenes superiores)
y de 2 cámaras (imágenes inferiores)
26Evaluación de la función cardiaca por imagen
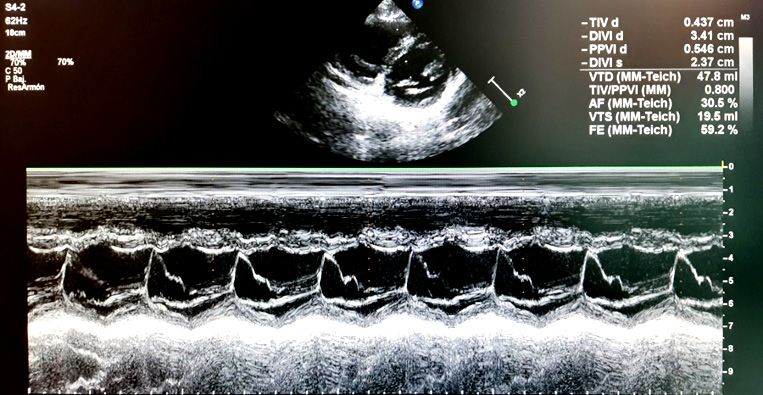
Figura 2. Cálculo de FA en modo M
En la Tabla 4 se muestran las fórmulas La función cardiaca se puede clasificar
de la FE y FA y los valores normales. según una escala cualitativa (Tabla 5):
Tabla 4. Fórmulas de la fracción de eyección Tabla 5. Escala cualitativa de disfunción
y fracción de acortamiento sistólica29
Fracción de eyección: Función sistólica del ventrículo izquierdo
FE% = (VDVI - VSVI) / VDVI × 100, Fracción de Fracción de
eyección acortamiento
donde VDVI: volumen diastólico del (FE) % (FA) %
ventrículo izquierdo; VSVI: volumen sistólico Hiperdinámica >70% >45%
del ventrículo izquierdo Normal 55-70% 26-45%
Disfunción 40-54% 20-25%
Valores normales FE: 55-70% leve
Fracción de acortamiento: Disfunción 30-39% 15-19%
FA% = (DDVI - DSVI) / DDVI × 100, moderada
DisfunciónTambién puede leer

















































