POLIARTERITIS NODOSA (PAN) KUSSMAUL Y MAIER (1866)
←
→
Transcripción del contenido de la página
Si su navegador no muestra la página correctamente, lea el contenido de la página a continuación
POLIARTERITIS NODOSA
(PAN)
KUSSMAUL Y MAIER (1866)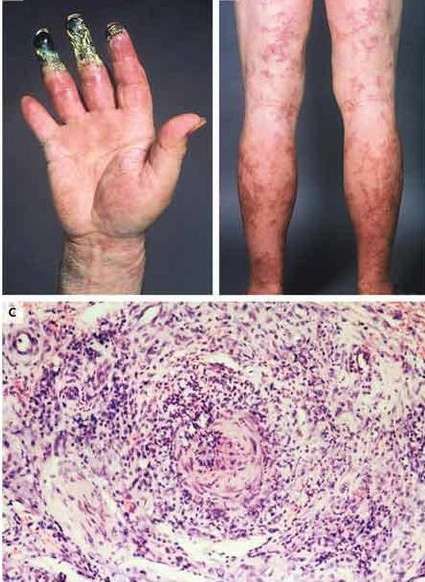
DEFINICIÓN: *Es una vasculitis necrotizante con manifestaciones sistémicas como consecuencia del compromiso de arterias musculares de pequeño y mediano calibre. *Es una inflamación de las paredes de los vasos generalmente acompañado de necrosis, no una inflamación alrededor de los vasos; si bien Kussmaul y Maier la llamaron Periarteritis Nodosa, es un proceso transmural, no perivascular, y actualmente se prefiere denominarla Poliarteritis nodosa o Panarteritis Nodosa. *La afectación vascular se caracteriza por ser segmentaria, con predilección por los sitios de ramificación y bifurcación de los trayectos vasculares. *Habitualmente respeta las arteriolas, vénulas y capilares, compromete arterias viscerales, pero respeta la circulación pulmonar.

EPIDEMIOLOGÍA La PAN es una enfermedad poco frecuente, en EEUU está incluida dentro de las enfermedades raras, menos de 20000 personas la padecen. La incidencia anual es de 0,7 / 100.000 y la prevalencia es de 6.3 / 100.000 habitantes. Afecta a hombres y mujeres en relación 1,6:1, con un rango de edad entre 40 y 60 años.
ETIOPATOGENIA
Se describen formas primarias de PAN.
Las formas secundarias son consecuencia de infecciones,
principalmente por el VHB, no obstante otros agentes
etiológicos han sido considerados responsables:
Virus de la hepatitis C Estreptococos
Virus de inmunodeficiencia humana Klebsiellas
Citomegalovirus Pseudomonas
Parvovirus B19 Yersinias
Virus linfotrópico T Humano Vacunación:
influenza,VHB y C
La asociación de PAN y VHB era del 15 y el 30%. La
vacunación contra este virus disminuyó la asociación al 10%.
En las poblaciones infectadas con VHB la prevalencia de PAN
es del 2-3%.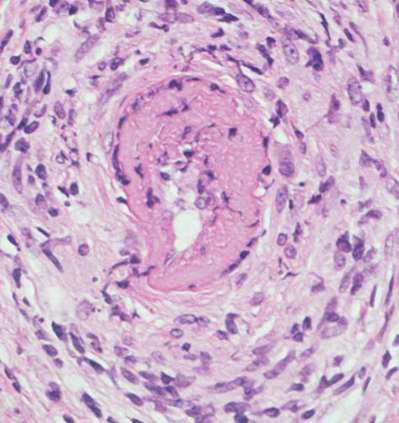
Se sugiere que la PAN es consecuencia del depósito, en la pared vascular, de inmunocomplejos solubles de tamaño superior a 19S, llevando a la activación de los componentes de la cascada del complemento, migración de leucocitos polimorfonucleares y consiguiente daño vascular. Se han observado LT CD4 + y macrófagos en el infiltrado que rodea los vasos musculares y perineurales, se infiere la participación de la inmunidad celular en la patogenia de la PAN.
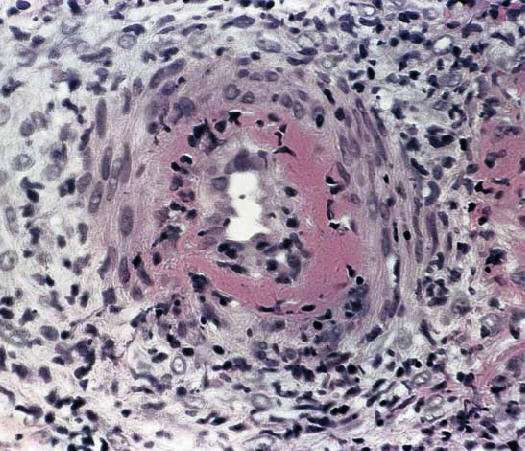
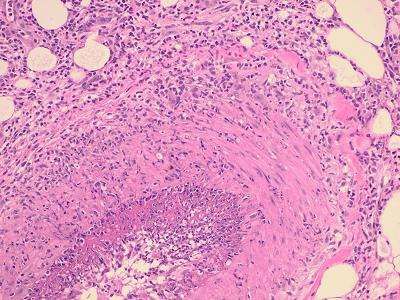
ANATOMÍA PATOLÓGICA La lesión característica de la PAN sistémica es una panarteritis de las arterias de pequeño y mediano calibre. El adjetivo nodosa indica que la alteración es focal y segmentaria, por lo tanto puede provocar tumefacciones nudosas. La degeneración de la pared arterial con depósito de material fibrinoide es la lesión aguda. El infiltrado inflamatorio es transmural (panarteritis), y está compuesto por PMN y Eo; se observa leucocitoclasia. En etapas ulteriores la proliferación de la íntima y la trombosis ocluyen la luz vascular, con isquemia, necrosis y ulceración. El compromiso sectorial puede dar origen a la formación de dilataciones vasculares de tipo microaneurisma. Aún cuando las arterias viscerales sufren las modificaciones típicas de la enfermedad, la participación de la piel, el TCS y los músculos esqueléticos se confina a vasos pequeños. A nivel cutáneo están alterados los vasos de la dermis profunda en la vecindad del TCS.
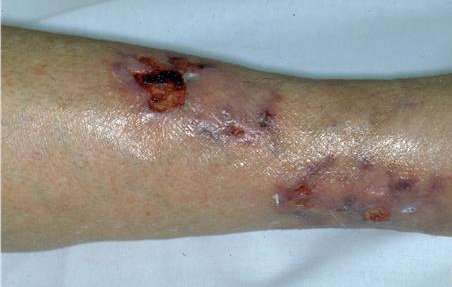
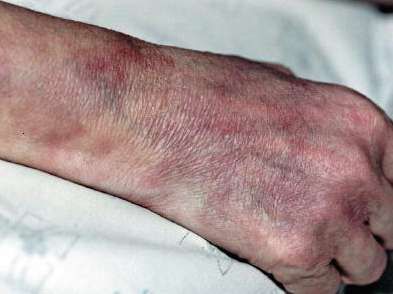
MANIFESTACIONES CLÍNICAS Son proteiformes, dependiendo del sector del árbol vascular comprometido. Síntomas generales: fiebre, adelgazamiento, astenia. Afección nefro-urológica: HTA, proteinuria, hematuria sin glomerulonefritis, cilindruria, insuficiencia renal. Afección neurológica: neuropatía periférica, ACV, convulsiones. Afección G-I: dolor abdominal, náuseas, vómitos, colecistitis, hemorragia digestiva, perforación, infarto intestinal. Vasculitis y aneurismas de los vasos del hígado. Afección cardíaca: insuficiencia cardíaca congestiva, cardiopatía isquémica, pericarditis. Compromiso músculo-esquelético: mialgias, artralgias, artritis oligoarticular de grandes articulaciones, sobre todo en miembros inferiores.
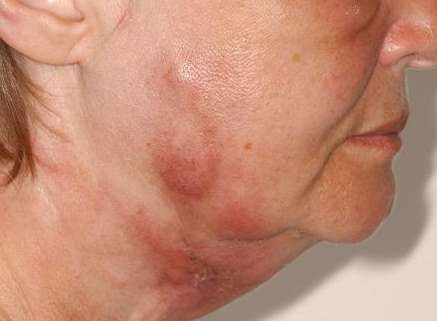
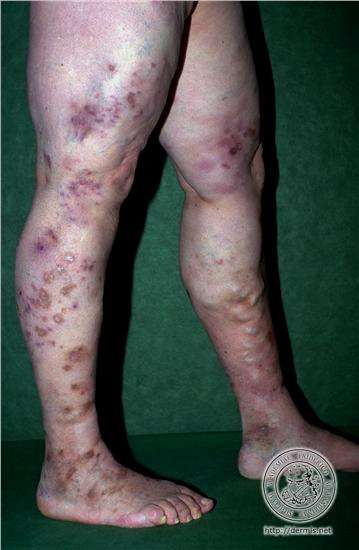
MANIFESTACIONES CUTÁNEAS Las lesiones cutáneas pueden ser el primer signo de la enfermedad, en general se limitan a los miembros inferiores, e incluyen púrpura y eritema. Las áreas purpúricas pueden contener ampollas y por infarto agudo, úlceras, asemejándose a una vasculitis leucocitoclástica grave, gangrena distal. Puede presentar, además, livedo reticular. El 20% de los pacientes presentan nódulos subcutáneos, expresión de aneurismas arteriales. Tienden a seguir el trayecto arterial, suelen ser dolorosos y la piel que los recubre presenta signos de inflamación.
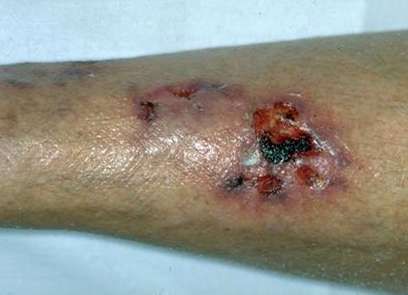
OTRAS MANIFESTACIONES: Orquiepididimitis. Alteraciones oculares. Compromiso pulmonar, (extremadamente raro): infiltrados en la Rx directa, derrame pleural.
PAN -VHB + La PAN-VHB+ podría considerarse una enfermedad por inmunocomplejos. Con el advenimiento de la vacuna con el VHB la entidad ha demostrado un descenso progresivo. Los adictos por vía e.v. han tomado un lugar preponderante como población de riesgo para PAN-VHB+. La PAN se presenta dentro de los 6 meses que siguen a la infección. El compromiso hepático es silente en la mayoría de los casos, y es la aparición de la PAN lo que obliga a la búsqueda de la infección viral. Las manifestaciones clínicas que la distinguen de la PAN idiopática incluyen: *pacientes menores de 40 años *HTA maligna *infartos renales *orquiepididimitis *compromiso G-I (emergencias quirúrgicas).
MÉTODOS COMPLEMENTARIOS LABORATORIO -Anemia(30%) -Leucocitosis -Trombocitosis>400.000/mm3 (50%) -VSG >20mm (90%) -Hematuria, proteinuria, cilindruria (50%) -Uremia > 60mg/dl (79%) -Hipoalbuminemia + hipergamaglobulinemia policlonal -Hipocomplementemia (30-70%) -Factor reumatoideo + (40%) -Crioglobulinas -FAN (-) p-ANCA (+) (10-20%) -HBsAg (30%). Actualmente < del 10%
BIOPSIA El diagnóstico de PAN se fundamenta en el hallazgo anatomopatológico de una vasculitis necrotizante de arterias de pequeño y mediano calibre (con necrosis fibrinoide), en un individuo con un cuadro clínico compatible. Los lugares más accesibles son la piel, músculo, testículo, en lugares clínicamente afectados. Para las biopsias percutáneas de hígado o riñón, deberá descartarse previamente la presencia de aneurismas por estudio angiográfico, para evitar el riesgo de sangrado intraparenquimatoso post punción.
ANGIOGRAFÍA Pone en evidencia la presencia de aneurismas y estenosis de vasos de mediano calibre. Los aneurismas miden entre 1-5mm y se encuentran en riñón, mesenterio e hígado. No son patognomónicos de PAN, dado que otras patologías pueden presentarlos: LES, Enfermedad de Kawasaki, Granulomatosis de Wegener, Endocarditis bacteriana, abuso de drogas, etc.
PAN. CRITERIOS DE CLASIFICACIÓN. (ACR 1990)
1. Pérdida de peso > de 4kg. (No por dietas u otros factores).
2. Livedo reticular.
3. Dolor testicular.
4. Mialgias, debilidad o dolor de miembros inferiores.
5. Mononeuropatía, mononeuritis múltiple o
polineuropatía.
6. Presión arterial diastólica > de 90 mmHg
7. Uremia > de 40mg/dl o creatininemia > de 1,5mg/dl
(no por deshidratación u obstrucción).
8. Evidencias de infección por VHB (presencia de Ag de
superficie o Ac séricos).
9. Arteriografía anormal (Aneurismas y/u oclusiones de arterias
viscerales).
10. Biopsia de arteria de pequeño o mediano calibre
con infiltrado inflamatorio de PMN (además de
mononucleares) en la pared de vaso.
Un paciente tendría PAN cuando al menos 3 de estos criterios están presentes.
Sensibilidad 82%; Especificidad 86%.FACTORES PRONÓSTICOS EN PAN
1. Compromiso Renal Proteinuria > de 3gr/24hs
Creatininemia > 1,58mg/dl
2. Compromiso G-I Sangrado gigestivo y/o
Perforación y/o
Infarto intestinal y/o
Pancreatitis.
3. Miocardiopatía
4. Compromiso del SNC
5. Pérdida de peso.TRATAMIENTO: El uso de corticoides mejoró la sobrevida del 13 al 48%, y la asociación con inmunosupresores como la ciclofosfamida , permitió supervivencias a 5 años del 80%. Dosis: Prednisona 1-2mg/kg/día. Cuando el estado clínico y métodos complementarios avalan una evolución favorable se comienza el descenso paulatino de la dosis, a partir del mes del inicio, hasta aquella que mantenga al paciente asintomático. Se puede utilizar el tratamiento días alternos. Se sabe que son varios años de terapéutica. Si el paciente muestras lesiones severas, con compromiso de vida, se deberá elegir la metilprednisona, en pulso endovenoso, 1gr/día por 3 días, para continuar con la dosis de prednisona oral.
Cuando la terapia esteroidea falla, los síntomas son de extrema gravedad, o hay riesgo de muerte, la indicación formal es el uso de drogas citotóxicas como la ciclofosfamida, dosis de 2mg/kg/día, vía oral. Efectos adversos: cistitis hemorrágica, fibrosis vesical cáncer de vejiga, supresión de médula ósea. Las infecciones representan la mayor causa de mortalidad, en pacientes con vasculitis sistémicas tratados con altas dosis de corticoides, asociados a drogas citotóxicas. La ciclofosfamida 1gr/m2 de superficie corporal, en pulsos endovenosos mensuales, por un año, produce una menor dosis acumulativa y expone al paciente a su toxicidad potencial por cortos períodos de tiempo. Alternativa: Azatioprina 2mg/kg/día, vía oral.
En casos refractarios a los tratamientos convencionales: *Plasmaféresis, 3 sesiones semanales por 3 sem. *Metotrexate 0,15-0,30mg/kg/semana. *Altas dosis de inmunoglobulina endovenosa: 2gr/kg durante 2 días, más prednisona oral, c/4-6 sem por 6 ciclos. *Mycofenolato mofetil 2gr/día, controló PAN en 2 niños, resistente a terapia convencional. *Anticuerpos monoclonales anti TNF alfa (Campath- 1)
Conducta terapéutica en pacientes PAN + VHB La PAN en pacientes VHB+ ocurre dentro de los primeros 6 meses que siguen a la infección viral, con compromiso renal, y alto nivel de replicación viral. Situación difícil: se debe revertir el proceso vasculítico y evitar la persistencia y replicación viral favorecida por los medicamentos inmunosupresores. La terapia corticoidea se asocia a antivirales: vidarabina o interferón alfa 2b + plasmaféresis.
Medidas de sostén: Control estricto de la presión arterial. Tratamiento farmacológico de la cardiopatía isquémica y de la insuficiencia cadíaca. Necesidad de hemodiálisis, nefrectomía, transplante renal. Prevención de neumonía por Pneumocystis Carinii en pacientes inmunosuprimidos: Trimetoprima sulfametozazol cuando el recuento de Lt CD4 sea menor de 300/mm3. Rehabilitación y mantenimiento del trofismo muscular por medios kinéticos.
FORMAS LIMITADAS DE PAN La PAN puede presentarse con compromiso exclusivo de determinados órganos sin afección sistémica. La vesícula, testículos, apéndice, SNP y piel se destacan entre las estructuras comprometidas. La PAN cutánea es una vasculitis crónica limitada a la piel, y en ocasiones se extiende a nervios, músculos y articulaciones adyacentes. Las lesiones son segmentarias pero a diferencia de la PAN sistémica no tiene preferencia por las bifurcaciones. Están comprometidas las arterias de pequeño y mediano calibre de la dermis reticular e hipodermis, tomando aquí la forma de una paniculitis septal con vasculitis. Se presenta como brotes de nódulos subcutáneos dolorosos, de color rojizo, de 5 a 20mm de diámetro, rodeados de livedo reticular. Aparecen con más frecuencia en las superficies extensoras de las piernas y menos frecuentemente en los brazos. Raramente siguen el curso de las arterias, la livedo reticular que los rodea le da la apariencia de “estallido estelar”. Estos nódulos se pueden ulcerar, sufrir hemorragia y necrosis, como resultado de la isquemia producida por la vasculitis.
El fenómeno de Raynaud puede acompañar la livedo reicular en las extremidades previo al desarrollo de los nódulos. El ANCA, FAN, factor reumatoideo, test serológico para hepatitis, crioglobulinas, hipocomplementemia, son negativos. Las lesiones pueden remitir en forma espontánea después de varias semanas sin dejar cicatrices, pero son comunes las recurrencias. Las exacerbaciones de esta enfermedad en general se acompañan de artralgias, neuropatía sensitiva periférica, mialgias y fiebre. Aunque dura meses, no se observa un compromiso sistémico grave. La PAN cutánea se ha asociado a infecciones estreptocócicas, hepatitis viral; en muchos casos no hay ninguna enfermedad subyacente.
Los nódulos cutáneos y subcutáneos de la PAN cutánea muestran el mismo cuadro histológico que la sistémica, es decir una panarteritis. Pueden identificarse distintos estadios en diferentes lesiones de un mismo paciente. En las recientes se aprecia degeneración de la pared arterial con depósito de material fibrinoide. Se produce la destrucción parcial o completa de las láminas elásticas externa e interna. El infiltrado ubicado en y alrededor de las paredes arteriales se compone de neutrófilos con manifestaciones de leucocitoclasia y algunos eosinófilos. En etapas ulteriores la proliferación de la íntima y la trombosis ocluyen la luz, esto causa isquemia y ulceración, el infiltrado incluye linfocitos y macrófagos. En la fase de curación se registra proliferación fibroblástica en el área perivascular.
Los estudios de inmunofluorescencia de la PAN cutánea revelan depósitos de Ig M y complemento en los vasos afectados. Un aumento moderado de la VSG y anemia son las alteraciones de laboratorio más frecuentes, con test inmunológicos negativos. El curso es fluctuante y es importante el seguimiento de los pacientes porque en algunos casos progresa a la forma sistémica. El tratamiento es difícil por el curso fluctuante de la enfermedad. En las formas moderadas se usan pequeñas dosis de prednisona (20mg/día) asociada a AINE; en las formas crónicas los antibióticos son de utilidad. En las formas severas el tratamiento es difícil, con inmunosupresores, y resultados controversiales, con persistencia de los síntomas por más de 20 años.
MUCHAS GRACIAS !!
También puede leer

















































