PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS - Genotipia
←
→
Transcripción del contenido de la página
Si su navegador no muestra la página correctamente, lea el contenido de la página a continuación
PATRONES DE TRANSMISIÓN DE
LAS ENFERMEDADES GENÉTICAS
MÓDULO 1: CONCEPTOS GENERALES DE GENÉTICA HUMANA
TÍTULO DE EXPERTO UNIVERSITARIO EN MEDICINA GENÉTICA Y GENÓMICA 2019
Material didáctico: Módulo 1-Clase 2
Material didáctico creado por Mar Benito, MSc https://www.linkedin.com/in/marbenito/
PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
MÓDULO 1: CONCEPTOS GENERALES DE GENÉTICA HUMANA
1.2.- PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS.
Profesor: Dr. Miguel Ángel García Pérez
ÍNDICE
1.- INTRODUCCIÓN. CONTRIBUCIÓN DE LOS GENES Y EL AMBIENTE…………………………......1
2- TIPOS DE ENFERMEDADES GENÉTICAS…………………………………………………………………......2
3-LAS ENFERMEDADES MENDELIANAS…………………………………………………………………..…......4
3.1- Características principales……………………………………………………………………..........4
3.2- Bases de datos………………………………………………………………………………….…..…......5
4.-PATRONES DE HERENCIA DE ENFERMEDADES MENDELIANAS……………………….….….......7
4.1.-Herencia Autosómica Dominante…………………………………………………………..…...10
4.2.-Herencia Autosómica Recesiva……………………………………………………………...…....13
4.3.-Herencia ligada a los cromosomas sexuales………………………………………………...16
5.-FACTORES QUE AFECTAN A LOS PATRONES DE HERENCIA BÁSICOS………………….…......20
5.1.- Penetrancia reducida................................................................................................................20
5.2.- Expresividad variable...............................................................................................................21
5.3.- Mutaciones de novo.......................................................................................................................23
5.4.- Mosaicismo germinal................................................................................................................23
5.5.- Genes con impronta genética.................................................................................................24
5.6.- Heterogenidad de locus............................................................................................................25
6.-HERENCIA MITOCONDRIAL……………………………………………………………………………….….....26
7.-PREGUNTAS.......................................................................................................................................................29
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 0
PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
1.- INTRODUCCIÓN. CONTRIBUCIÓN DE LOS GENES Y DEL AMBIENTE
Durante los últimos años, los avances en el campo de la genética han transformado la
medicina, así como la forma de entender muchas de las enfermedades que afectan a los
humanos. Desde la identificación de los primeros genes responsables de enfermedades
hereditarias, en la época de los años 80, y con la secuenciación del genoma humano, el
conocimiento sobre las bases moleculares de algunas de estas enfermedades humanas se
ha visto incrementado de forma exponencial. Además, conocemos la gran complejidad de
nuestro genoma y de los mecanismos de regulación, lo que nos indica que la relación entre
el fenotipo y el genotipo no es tan simple como se pensaba. Esto se puede extrapolar a las
enfermedades con componente genético, así la relación entre el genotipo y las
manifestaciones clínicas es bastante compleja. Los genes, además de codificar para
proteínas también pueden codificar ARNs funcionales, con un importante papel de
regulación, que se expresan en momentos y tejidos concretos y que funcionan en
colaboración con otras proteínas formando redes bioquímicas o rutas del desarrollo. En el
genoma humano hay más de 20.000 genes que se expresan, de estos 2 terceras partes
sufren splicing alternativo, lo que da lugar a casi unas 10.000 proteínas. Además, muchas
de estas proteínas sufren modificaciones postraduccionales. No todos los tejidos están
expresando todo el genoma, el transcriptoma de cada uno contiene unos 5000 ARN
mensajeros. Las proteínas también interactúan entre ellas formando redes. Todo esto da
lugar a una gran variabilidad fenotípica.
En la actualidad, existen nuevos proyectos que continúan profundizando en este
conocimiento, como el HapMap o el proyecto de los 1000 genomas, que estudian la
variabilidad genética de las poblaciones humanas, y el proyecto ENCODE, enfocado en
identificar los elementos funcionales de la secuencia del genoma humano.
La identificación de los genes responsables de enfermedades hereditarias o que pueden
ser factores de riesgo, supone un paso esencial, no sólo para conocer cuáles son los
mecanismos fisiopatológicos de dichas enfermedades, esto es, cuál es la función del gen
responsable y cómo afecta la mutación a la misma, sino también para poder diseñar las
pruebas necesarias para el diagnóstico genético y los fármacos para su tratamiento.
La relación entre un gen y una enfermedad no es simplista. A nivel general, se puede
considerar que todas las enfermedades que afectan a los seres humanos son el resultado
de la combinación de los genes y el ambiente. Sin embargo, la contribución de los genes en
cada una de las enfermedades puede ser diferente. Hay enfermedades en las cuales los
factores genéticos tienen un impacto muy fuerte en el fenotipo, representando el factor de
riesgo más importante, como por ejemplo la fibrosis quística o la hemofilia. En cambio, en
otras enfermedades, la contribución del medio ambiente es más significativa,
constituyendo el principal factor de riesgo, como por ejemplo en cáncer de pulmón. El caso
extremo lo constituyen los accidentes de tráfico, que tienen un componente totalmente
ambiental. Entre ambos extremos, se encuentra una gran variabilidad de enfermedades
con diferente contribución genética y ambiental.
Por tanto, es importante conocer qué factores, genéticos o ambientales, tienen un mayor
peso en el desarrollo de una enfermedad determinada, para poder actuar en consecuencia
a través de políticas de salud pública o de atención a los pacientes. En el ejemplo del
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 1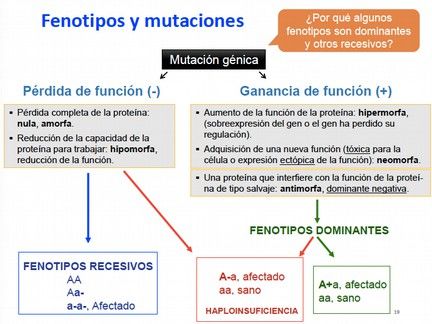
PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
cáncer, tenemos cánceres como el de mama, provocados por mutaciones en los genes
BRCA1 y BRCA2, con un importante componente genético, y cánceres como el de pulmón
asociado al consumo de tabaco, que es un factor ambiental.
De esta manera, si el factor de riesgo más importante es un único gen, se deberá
recomendar un seguimiento exhaustivo de los miembros de la familia del enfermo para
determinar quiénes son susceptibles de desarrollar la enfermedad. Mientras que si el
factor de riesgo más importante es ambiental, se realizarán acciones para concienciar a la
población sobre la utilización de determinados compuestos.
En cada vértice del triángulo de la figura de la diapositiva 3 podemos situar un tipo de
enfermedad. En un vértice se sitúan las enfermedades monogénicas o mendelianas, en las
que mutaciones en genes individuales constituyen el factor más importante. En otro
vértice se encontrarían las enfermedades en las que el factor ambiental es clave. Y en el
tercero, las enfermedades en las que contribuye más de un gen (poligénicas), y que además
están muy influenciadas por el ambiente. Cada enfermedad que nos afecta se podría situar
en un punto distinto del triángulo.
DIAPOSITIVAS 2 Y 3
2.-TIPOS DE ENFERMEDADES GENÉTICAS
Se consideran enfermedades genéticas aquellas en las que existe un componente genético
o hereditario implicado. Se clasifican en cromosómicas, monogénicas y multifactoriales o
de herencia compleja.
Las alteraciones cromosómicas o cromosopatías, que afectan en su conjunto a 7 de cada
1.000 nacimientos, son debidas a alteraciones en el número o estructura de los
cromosomas. Estas alteraciones cromosómicas son responsables de aproximadamente el
50% de los abortos espontáneos del primer trimestre, y pocos de los embriones que las
contienen llegan a término. El síndrome de Down (trisomía del cromosoma 21), el
síndrome del Cri du Chat (deleción del brazo corto del cromosoma 5) y el síndrome de
Turner (monosomía del cromosoma X), son algunos ejemplos de enfermedades
cromosómicas.
Las enfermedades monogénicas (también llamadas mendelianas) son aquellas causadas
por mutaciones en genes individuales. Estas enfermedades presentan patrones específicos
de transmisión y tienen una prevalencia de 2 casos sobre 100 en la población general. Si
las consideramos de forma individual son enfermedades raras, pero en su conjunto son
enfermedades comunes (3-8% en la población general). La fibrosis quística o la
enfermedad de Huntington son ejemplos de alteraciones en genes nucleares, mientras que
el síndrome de Kearns-Sayre es un ejemplo de mutación en un gen mitocondrial.
L a s enfermedades multifactoriales (o de herencia compleja) son aquellas en las que
contribuyen a la enfermedad tanto factores genéticos como factores de tipo ambiental, o la
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 2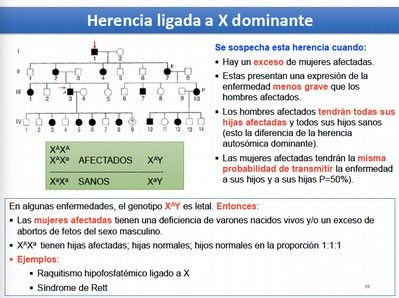
PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
interacción entre ambos. Son las enfermedades hereditarias más numerosas, y el
porcentaje de afectados va aumentando a medida que se van encontrando nuevos genes
responsables de enfermedades ya conocidas, por ahora está en un 60% de la población. Se
consideran responsables de muchas de las malformaciones congénitas (labio leporino o
alteraciones en el tubo neural), así como de enfermedades propias de la población adulta,
como por ejemplo la diabetes de tipo 2, hipertensión, cardiopatías, numerosas
enfermedades psiquiátricas, etc.
Como se ve en el gráfico de la diapositiva 5, muchas alteraciones en el cariotipo no son
compatibles con la vida, y por tanto se dan abortos espontáneos. El síndrome de Down es
un ejemplo de alteración cromosómica en que los afectados pueden llegar a adultos. Aquí
la esperanza de vida ha aumentado mucho, desde los 12 años de mitad del siglo pasado a
los 60 años en la actualidad.
Algunas de las enfermedades mendelianas ya se diagnostican en el nacimiento, y otras
debutan en la fase adulta.
Las enfermedades multifactoriales son responsables de muchas alteraciones congénitas
pero también pueden aparecer en la fase adulta.
Una mutación es cualquier cambio en la secuencia de ADN o alteración cromosómica, pero
en genética médica, se habla de mutaciones cuando estos cambios tienen un efecto
patológico.
Las enfermedades monogénicas son las únicas que presentan un patrón de herencia claro,
ya que al estar causadas por mutaciones en un único gen, suelen transmitirse a la
descendencia siguiendo un patrón de herencia mendeliano, que puede rastrearse en la
familia afectada. En estos casos la mutación es rara pero tiene un fuerte impacto en el
fenotipo, la penetrancia es alta, es decir el riesgo de desarrollar la enfermedad será alto, y
además será igual para todas las familias que presenten esa mutación. Para el estudio de
estas mutaciones se utiliza la secuenciación.
En las enfermedades de herencia compleja, diferentes genes contribuyen a la enfermedad,
de forma que la mutación en uno de esos genes no es suficiente para que se manifieste la
enfermedad. Aquí se utiliza el concepto de variación génica o polimorfismo. En este caso,
para que un individuo desarrolle la enfermedad debe de tener una combinación
determinada de los genes implicados. De esta manera, las mutaciones en genes
individuales pueden tener una frecuencia alta en la población sin causar un efecto en el
fenotipo, tienen poca penetrancia. Todo ello, sin olvidar la contribución del ambiente. Por
tanto, en las enfermedades complejas es más difícil rastrear las mutaciones a partir de la
genealogía de una familia, y los patrones de herencia no están claros. Cada mutación puede
tener una contribución fenotípica diferente, además, el riesgo para cada familia puede ser
distinto en función del conjunto de factores de riesgo que presente.
Para el estudio de las enfermedades de herencia compleja se llevan acabo los estudios de
asociación (GWAS), en los que se mira para toda una batería de genes distribuidos por
todo el genoma, si ciertos polimorfismos son más frecuentes en los individuos afectados
que en los controles. Lo que se obtiene es un resultado estadístico, y se establece la
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 3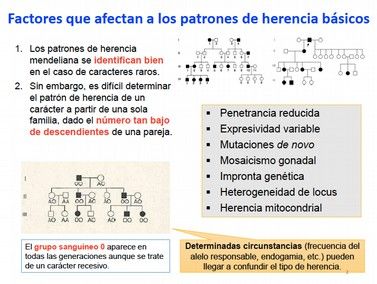
PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
hipótesis de que ciertos polimorfismos relacionados con los pacientes podrían ser los
causantes de la enfermedad.
Como se ve en la diapositiva 7 las mutaciones que dan lugar a enfermedades monogénicas
son muy poco frecuentes en la población, por ello estas enfermedades son raras, pero el
efecto de las mutaciones sobre el fenotipo es muy importante. Las enfermedades
multifactoriales son más comunes, pero el efecto de las mutaciones en el fenotipo es más
bajo.
También hay variantes raras con poco efecto en la población, pero son muy difíciles de
detectar. Y variantes comunes con mucho efecto sobre la población, pero son muy pocas.
Polimorfismo en el gen 5HTT como ejemplo de herencia compleja
La región reguladora del gen 5HTT, que codifica para el receptor de la serotonina contiene
un polimorfismo consistente en la inserción o deleción de un fragmento de 44 pares de
bases que da lugar a dos alelos, alelo corto o S, con 14 repeticiones y alelo largo o L con 16
repeticiones. El alelo S se ha asociado a una mayor probabilidad de depresión en función
de ciertos componentes ambientales. Los tres genotipos posibles no se diferencian cuando
las experiencias o vivencias son normales. Sin embargo, en presencia de acontecimientos
vitales graves, como la pérdida de un familiar, o la pérdida del trabajo, se observan
diferencias entre las personas con diferente genotipo.
En las enfermedades monogénicas una mutación es necesaria y suficiente para desarrollar
ella enfermedad. En estas familias se puede seguir la enfermedad en el árbol genealógico.
La mutación suele ser responsable del 100% del fenotipo. Aunque hay que tener en cuenta
la penetrancia y el background genético, ya que puede haber genes modificadores de los
que tienen la mutación. La mutación en diferentes familias tiene el mismo efecto.
En las enfermedades complejas hablamos de variantes genéticas o alélicas. Cada una de
estas variantes contribuye muy poco al fenotipo final, tienen un efecto aditivo. En este caso
los árboles genealógicos son complejos y difíciles de interpretar. Se detecta una agregación
familiar, porque las familias comparten genes y ambiente. El efecto en cada familia es
distinto, cada variante tiene un efecto diferente en cada caso.
DIAPOSITIVAS 4-7. Enfermedades genéticas
3.-LAS ENFERMEDADES MONOGÉNICAS
3.1.- Características principales
Las enfermedades monogénicas son debidas a alteraciones o mutaciones en un único gen,
localizado, bien en el núcleo o bien en las mitocondrias. Las mutaciones en genes nucleares
presentan una herencia típicamente mendeliana, es decir, siguen las leyes de transmisión
de la herencia de Mendel, mientras que las mutaciones en genes mitocondriales presentan
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 4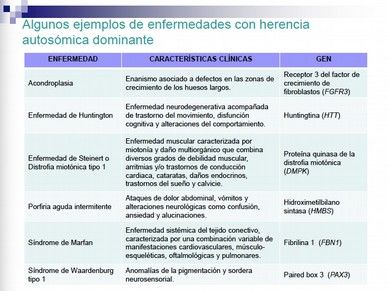
PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
una herencia materna, de modo que la transmisión de la enfermedad se produce
exclusivamente a través del linaje materno, ya que son las madres las que contribuyen con
las mitocondrias.
Enfermedades mendelianas
Individualmente, las enfermedades mendelianas son poco frecuentes. Sin embargo,
globalmente estas enfermedades afectan a un 2% de la población, con algunas
estimaciones de la Organización Mundial de la Salud de hasta un 6-8% de la población,
originadas por el aumento en la identificación de genes responsables de patologías
humanas de baja frecuencia.
Se entiende por Enfermedad Rara aquella enfermedad cuya incidencia es menor de 1
casos por cada 2.000 individuos. Según la UE son: “ Enfermedades, incluidas las de origen
genético, que son crónicamente debilitantes o potencialmente mortales y las cuales tienen
tan poca prevalencia que se necesitan esfuerzos especiales combinados para combatirlas”.
En la actualidad, se han descrito alrededor de 7.000 enfermedades raras distintas, de las
que el 80% son genéticas, pero una proporción significativa sigue sin haberse
caracterizado de forma molecular. En España se estima que hay 3 millones de afectados.
Estas enfermedades suelen afectar a las capacidades físicas, habilidades mentales,
cualidades sensoriales o de comportamiento. Muchas son crónicas, degenerativas e
incapacitantes.
Además, los recursos terapéuticos disponibles para el tratamiento de las enfermedades
raras son bastante limitados. En este contexto, surge el concepto de medicamento
huérfano, como aquel fármaco destinado a diagnosticar, prevenir o tratar una enfermedad
rara. La baja prevalencia en la población de estas enfermedades hace que las grandes
compañías farmacéuticas no muestren un interés especial en desarrollar medicamentos
huérfanos, ya que el número de personas que se beneficiarían del tratamiento puede no
compensarles el esfuerzo económico para su desarrollo. Este problema ha requerido la
intervención de los gobiernos, quienes incentivan a las empresas farmacéuticas, para que
desarrollen medicamentos huérfanos, con ventajas fiscales o económicas en forma de
subvenciones.
DIAPOSITIVAS 8 Y 9. Enfermedades mendelianas
3.2.- Bases de datos de caracteres mendelianos
OMIM (http://www.ncbl.nlm.nih.gov/Omlm)
OMIM (Online Mendelian Inheritance in Man), es la principal base de datos de los
caracteres mendelianos humanos. Se generó por primera vez en los años 60 en forma de
libro y actualmente se encuentra disponible online, bajo la forma de un catálogo de los
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 5
PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
genes humanos y de los trastornos humanos genéticos, la mayoría de ellos mendelianos,
aunque también incluye algunas enfermedades complejas y de herencia mitocondrial.
En la diapositiva 10 se muestran las estadísticas actualizadas (11 febrero 2019) de esta
base de datos. Hay unas 25.000 entradas de genes y fenotipos.
Al realizar una búsqueda para una enfermedad concreta, la base de datos devuelve una
serie de entradas que incluyen información sobre el fenotipo de la enfermedad, el patrón
de herencia, una revisión bibliográfica de los estudios realizados en la enfermedad y otros
datos de información relevantes. Las patologías con el símbolo # son las mejor conocidas.
Si no tiene símbolo significa que es una patología que no se sabe seguro si tiene una
herencia mendeliana.
Ejemplo:
Si introducimos el término “Cystic fibrosis”, esta búsqueda nos devuelve el código OMIM
#219700, donde el símbolo # nos indica que es una descripción fenotípica de base
molecular conocida, y el número 2 nos indica que tiene un patrón de herencia autosómico
recesivo. El número 1 indicaría que es autosómica dominante, el 3 que es ligada al
cromosoma x, el 4 ligada a cromosoma Y, el 5 herencia mitocondrial y el 6 son
enfermedades descubiertas después de 1994. Otra información hace referencia a las
características clínicas de los pacientes, los modos de herencia, si se han hecho estudios
citogenéticos, dónde se localizan las mutaciones, cómo se ha llegado a localizar el gen,
cómo las distintas mutaciones afectan a la función del gen, los aspectos bioquímicos
funcionales de la enfermedad, cómo se diagnostica, cómo se trata, la frecuencia de las
distintas mutaciones en la población, y la evolución. También nos da información de
fenotipos asociados y de genes modificadores
Otros datos de interés que se presentan en la base de datos son la correlación fenotipo-
genotipo según la mutación, los modelos animales descritos, un poco de historia y una
gran relación de bibliografía a la que se puede acceder directamente pinchando en los
enlaces correspondientes.
GENECARDS (http://www.genecards.org/)
Esta base de datos proporciona información sobre la función biológica de los genes ya
descritos y de los predichos, genómica, transcriptómica, etc.. También informa de si hay
modelos animales o anticuerpos para el gen, herramientas importantes para la
investigación.
GENETESTS (http://www.ncbi.mih.gov/sites/GeneTests/)
Proporciona información clínica de las enfermedades genéticas e incluye un directorio de
laboratorios donde se realizan o producen diferentes pruebas genéticas para unas 3000
enfermedades hereditarias, así como un directorio de lugares donde se puede obtener
asesoramiento relacionado con las enfermedades humanas hereditarias.
ORPHANET (http://www.orpha.net/consor/cgi-bin/index.php?lng=ES)
Es el portal de referencia para las enfermedades raras y los medicamentos huérfanos en
Europa. Hay unas 6000 enfermedades. En la diapositiva 12 se especifica toda la
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 6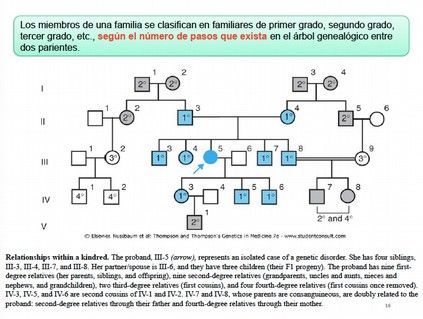
PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
información que se puede obtener en este portal. En algunos casos hay revisiones de la
enfermedad hechas por especialistas. También hay herramientas de soporte del
diagnóstico para los médicos, en las que se pueden introducir los síntomas y averiguar cual
es la posible enfermedad, y directorios de laboratorios clínicos, etc. Además, cada 15 días
aparece un boletín divulgativo de los avances en las enfermedades raras.
FEDER (http://web.enfermedades-raras.org)
Es la Federación Española de Enfermedades raras, impulsada por pacientes y afectados. Su
objetivo es luchar para los pacientes que son pocos para cada enfermedad individual, tanto
para los que tienen un diagnóstico como para los que todavía no lo tienen.
CIBERER: (http://www.ciberer.es)
El CIBER (Centro de investigación biomédica en red) de Enfermedades Raras es uno de los
nueve consorcios públicos establecidos por iniciativa del Instituto de Salud Carlos III,
creado para servir de referencia, coordinar y potenciar la investigación sobre las
enfermedades raras en España. Está formado por 62 grupos de investigación, ligados a 30
instituciones.
DIAPOSITIVAS 10-13 Bases de datos
4.-PATRONES DE HERENCIA DE ENFERMEDADES MENDELIANAS
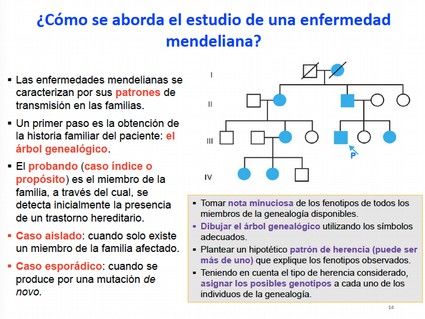
Para abordar el estudio de una enfermedad mendeliana, el primer paso es obtener la
historia familiar del paciente, esto es construir un árbol genealógico de la familia para
establecer quiénes son los individuos afectados y su relación entre ellos. El objetivo es
disponer de la mayor información posible sobre la transmisión de la enfermedad.
En la elaboración de árboles genealógicos la nomenclatura utilizada es la siguiente:
Caso índice, probando o propósito: es la persona de la familia con la que se inicia el
estudio, es decir el miembro de la familia a través del cual se detecta inicialmente la
presencia de un trastorno hereditario.
Caso aislado: se utiliza cuando en la familia hay un único miembro afectado.
Caso esporádico: se utiliza cuando la mutación se produce de novo, de forma que ninguno
de los progenitores tiene la mutación, y esta se ha producido en la línea germinal de uno
de ellos de forma espontánea. Suelen ser mutaciones de herencia dominante.
Hay que tomar nota minuciosa de los fenotipos de todos los miembros de la familia
disponibles. Dibujar el árbol genealógico. Plantear un hipotético patrón de herencia que
explique los fenotipos observados. Asignar los posibles genotipos a cada uno de los
individuos de la genealogía.
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 7
PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
En la elaboración de los árboles genealógicos se utiliza un lenguaje de símbolos (ver
diapositiva 15) para referirse, bien al sexo, bien al carácter enfermo, sano o portador o
bien al tipo de relación familiar entre los individuos. Cada generación se indica con
números romanos, y dentro de esta, cada individuo se representa con números arábicos
según el orden de nacimiento. Esta nomenclatura está unificada, con el objetivo de que
pueda ser utilizada a nivel internacional.
Los individuos con una raya vertical dentro son individuos que en ese momento no
manifiestan la enfermedad, pero que puede que lo hagan en el futuro. Los individuos con
un punto dentro significa que son portadores de una enfermedad recesiva y que por tanto
no van a manifestar el fenotipo. Este puntito no se pone solo en patologías ligadas al X.
Una vez construido el árbol genealógico, se pueden ver las relaciones de parentesco entre
los distintos miembros con respecto al caso índice. Los diferentes miembros de una familia
se clasifican en familiares de primer grado (padres, hijos y hermanos; indicados en azul en
diapositiva 16), de segundo grado (abuelos, nietos, sobrinos y tíos,…; indicados en gris en
diapositiva 16), de tercer grado (primos), y así sucesivamente, según el número de pasos
generacionales entre dos parientes que exista en el árbol genealógico. Esto permite
calcular el porcentaje de genoma compartido entre los diferentes individuos.
DIAPOSITIVAS 14-16
Los Patrones de Herencia en las enfermedades mendelianas, se definen según dos
criterios:
1. Localización del gen:
Si el gen responsable se localiza en cualquiera de los autosomas se trata de herencia
autosómica, y no habrá diferencia entre hombres y mujeres. Mientras que si se
localiza en los cromosomas sexuales se trata de herencia ligada a los cromosomas
sexuales.
2. La relación entre los alelos:
Se refiere a la relación entre el alelo mutado y el alelo normal o funcional, que puede
ser dominante o recesiva. Para saber cuál es esta relación, se observan los individuos
heterocigotos, los cuales presentan ambos alelos, el mutado y el normal. Si el individuo
heterocigoto que presenta la mutación en uno de los alelos está enfermo, se tratará de
u n a herencia dominante, mientras que si está sano, se tratará de una herencia
recesiva (ver diapositiva 17).
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 8PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
En los árboles genealógicos se diferencia entre alelo dominante y recesivo mediante la
utilización de letras mayúsculas (para los alelos dominantes) y minúsculas (para los
alelos recesivos).
Atendiendo a estos criterios, existen 5 patrones de herencia básicos:
- (A) Herencia Autosómica Dominante
- (B) Herencia Autosómica Recesiva
- (C) Herencia ligada al cromosoma X recesiva
- (D) Herencia ligada al cromosoma X dominante
- (E) Herencia ligada al cromosoma Y, los casos que se han encontrado se sospecha
que realmente son de herencia autosómica. Los genes del cromosoma Y controlan el
desarrollo de la gónada masculina y la formación de los gametos, por lo que las mutaciones
en estos genes no se pueden transmitir.
- Herencia parcialmente ligada al sexo o pseudoautosómica
La razón de que unas mutaciones presenten una herencia recesiva mientras que otras
presentan una herencia dominante, depende de cómo afecta la mutación a la función del
gen y cómo de importante es esta función. Esto determinará si la mutación se manifestará
y si alterará o no el fenotipo del individuo:
- Pérdida de función (-): Se produce cuando el producto del gen afectado no se
sintetiza (nula o amorfa), la cantidad producida está disminuida, o en caso de sintetizarse,
hay una reducción de la función (hipomorfa). Un ejemplo se produce cuando un gen está
delecionado, o ausente. Este tipo de mutaciones suelen dar fenotipos recesivos, porque a
pesar de que la presencia de mutación en uno de los alelos conlleva que ese alelo no
produzca la proteína correspondiente, los heterocigotos todavía tienen el 50% del
producto obtenido del alelo normal, que suele ser suficiente. Así, solo los homozigotos
presentan la enfermedad.
Como excepción, hay casos donde la dosis de un producto es crítica, como en los genes
cuyos productos presentan cantidades estequiométricas fijas con productos de otros
genes, y cualquier alteración modificaría las proporciones. Un ejemplo de este fenómeno,
denominado haploinsuficiencia (una sola dosis del alelo normal no es suficiente para un
fenotipo normal), se produce en el caso de la producción de hemoglobina, formada por dos
cadenas proteicas alfa y dos cadenas proteicas beta. Si hay una disminución de las cadenas
alfa, aumentará relativamente la concentración de cadenas beta, lo que podría derivar en el
ensamblaje de homotetrámeros de beta globina que son insolubles, precipitan y dañan a
los glóbulos rojos, produciendo anemia. Otro ejemplo son los genes que compiten entre
ellos para que se inicien ciertas rutas del desarrollo. Según la expresión de un gen respecto
del otro se iniciará una ruta u otra.
- Ganancia de función (+): Se produce cuando debido a la mutación se sintetiza una
nueva proteína que es tóxica para la célula, que realiza una función distinta, o que se
expresa en un lugar y tiempo inadecuado (neomorfa), o en casos donde la mutación da
lugar a una sobreexpresión del gen o a la pérdida de regulación (hipermorfa). También
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 9PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
puede ocurrir que la nueva proteína interfiera con la función de la proteína de tipo salvaje
(antimorfa). Estas mutaciones también se denominan dominantes negativas. Por ejemplo,
en proteínas que forman dímeros pueden ser que un monómero inactivo se una con otro
activo, y no le deje realizar su función.
DIAPOSITIVAS 17-19
4.1.-Herencia Autosómica Dominante
Este tipo de herencia afecta a 7 de cada 1000 individuos y representa más del 50% de
todos los trastornos mendelianos conocidos. La principal característica de la herencia
autosómica dominante es que se encuentran individuos afectados en todas las
generaciones (debido al carácter dominante), afecta a ambos sexos por igual (debido a que
la mutación está en un autosoma) y tanto los hombres como las mujeres tienen la misma
probabilidad de transmitirla a sus hijos de ambos sexos. Además, si en una rama
desaparece la mutación, ya no se transmitirá más dentro de la descendencia de la misma.
Es poco frecuente que los individuos afectados sean homocigotos, debido a la baja
frecuencia de los alelos mutantes en la población, y la poca probabilidad de que la pareja
esté formada por dos individuos afectados Pero si esto ocurre, la pareja de afectados puede
tener un hijo sano. Como excepción, se encuentra la enfermedad acondroplasia (enanismo
asociado a defectos en las zonas de crecimiento de los huesos largos) en la que los
afectados sí suelen formar pareja entre ellos. Otra razón para explicar la baja frecuencia de
homocigotos es que en muchos casos la condición es letal y provoca abortos espontáneos.
La probabilidad de transmisión de una persona afectada a sus hijos es del 50%. Si la
persona está sana, no tendrá riesgo de transmitir la enfermedad a la descendencia. El
riesgo de recurrencia, es decir la probabilidad de que teniendo un hijo afectado, el
siguiente también lo sea, es también del 50%, al tener en cuenta que cada hijo es un suceso
independiente.
Cuanto menor es la capacidad reproductiva debida a la mutación, mayor es la proporción
de casos debidos a mutaciones nuevas.
En el dominante puro el AA y el Aa tienen el mismo fenotipo. Esto no es muy frecuente, se
da en la enfermedad de Huntington. En el dominante incompleto o herencia incompleta
dominante, el fenotipo AA es más grave que el Aa. Esto se da en la hipercolesterolemia
familiar.
Ejemplos de enfermedades que se presentan con herencia autosómica dominante
(diapositiva 21):
Acondroplasia. Causada por una ganancia de función en el receptor 3 del factor de
crecimiento de fibroblastos, que queda activado constitutivamente, lo que hace que
los condrocitos no se desarrollen como toca.
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 10PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
Hipercolesterolemia familiar, es una de las más frecuentes.
Enfermedad de Huntington
Porfiria aguda intermitente. Se da una pérdida de función por haploinsuficiencia.
La mutación está en el gen HMBS que pertenece a la ruta de síntesis del grupo
hemo.
Síndrome de Marfan. Está causado tanto por pérdida de función por
haploinsuficiencia como por mutaciones dominantes negativas. Es un ejemplo de
mutación pleiotrópica, es decir, que afecta a muchos sistemas y órganos distintos.
DIAPOSITIVAS 20 Y 21 Herencia autosómica dominante
Enfermedad de Huntington como ejemplo de enfermedad autosómica dominante
Al buscar la enfermedad en OMIM obtenemos la entrada #143100. El 1 significa que es
autosómica dominante.
Esta enfermedad fue descrita en 1872 por George Huntington. Sus síntomas son:
Trastorno del movimiento (corea).
Disfunción cognitiva (lentitud de pensamiento, dificultad en recordar, deterioro en
las funciones intelectuales).
Alteraciones del comportamiento /psiquiátricas (depresión, irritabilidad,
impulsividad, etc.)
Late onset disease. Los síntomas se presentan entre la tercera-cuarta década de la
vida (media en 35-51 años). La supervivencia media es de 18 años después del
diagnóstico.
Variante Juvenil (5-10% de los casos) aparece en las dos primeras décadas de vida.
Forma más grave.
Variante senil (25% de los casos) debuta alrededor de los 50 años. La progresión
de la enfermedad es más lenta.
Al ser una enfermedad de aparición tardía, las personas que desarrollan la enfermedad,
frecuentemente han tenido hijos antes de saber que llevan el defecto génico. El retraso en
la edad de aparición de la enfermedad, reduce el efecto de la selección natural contra el
gen defectivo, explicando su frecuencia en la población, aunque se trate de un gen con una
de las tasas de mutación más bajas que se conoce.
La enfermedad de Huntington es debida a la mutación en el gen HTT, que codifica para la
proteína Huntingtina. La mutación responsable es debida a la expansión anómala de una
repetición del trinucleótido CAG, localizada en el primer exón del gen. Al analizar el
tamaño de las repeticiones entre el alelo normal y el mutado, se observa que los alelos en
la población normal (la mayoría entre 15-16) presentan muchas menos repeticiones que
los enfermos (la mayoría con alrededor de 45, aunque pueden alcanzar más de 100).
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 11PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
Existe una zona de solapamiento entre la población normal y los pacientes afectados con la
enfermedad para el número de repeticiones, de forma que el diagnóstico de las personas
con ese número de repeticiones puede ser ambiguo. Las personas que tienen entre 36 y 39
repeticiones la penetrancia no es completa pero tienen muchas probabilidades de
transmitir la enfermedad a la siguiente generación, ya que el número de repeticiones es
inestable y puede aumentar en la descendencia. Debido a esto se da el fenómeno de la
anticipación, los hijos de los afectados cada vez presentan la enfermedad a edades más
tempranas. A partir de 40 repeticiones la penetrancia es completa. De forma general, hay
formas juveniles con más de de 60 repeticiones y formas del adulto con 36-50
repeticiones.
Existe una variación poblacional de la enfermedad de Huntington, se presenta con mayor
frecuencia en ciertas poblaciones, como la europea (1 de cada 10.000) (efecto fundador), y
con menor frecuencia en las poblaciones asiáticas o africanas. Esto se debe al número de
repeticiones de los alelos normales en dichas poblaciones. En la población europea
abundan los alelos normales que presentan ya un número de repeticiones elevado, y por
tanto tienen una mayor probabilidad de expandirse superando el umbral patológico
durante la formación de los gametos, por lo que se transmitirá a la descendencia
(diapositiva 24). Este tipo de enfermedades con expansión de tripletes en zonas
codificantes suelen estar producidas por fallos en la meiosis de la gametogénesis paterna,
mientras que las enfermedades con expansiones en zonas no codificantes (ataxia de
Friedrich) suelen estar causadas por fallos en la gametogénesis materna.
Se pudo determinar el gen implicado en la enfermedad gracias a una familia venezolana de
18.000 individuos en la que se pudo llegar a ver que la primera portadora fue una mujer
con antecedentes españoles.
La Huntingtina es una proteína ubicua, que se expresa en muchos tejidos y localizaciones
subcelulares, además de interaccionar con otras proteínas celulares. Aunque se desconoce
la función exacta de la proteína normal, se la asocia con muchas funciones celulares como
el tráfico de proteínas, transporte de vesículas y anclaje al citoesqueleto, participación en
la sinapsis, apoptosis, control de la expresión génica, etc. Las células más sensibles a la
mutación en la Huntingtina son las neuronas espinosas de tamaño mediano, muy
abundantes en el cuerpo estriado del cerebro.
El triplete CAG en la región repetitiva del exón 1 del gen de la huntingtina corresponde al
aminoácido glutamina en la proteína. Cuando se supera el umbral patológico de
repeticiones, se obtiene un fragmento de glutaminas demasiado largo, lo que da lugar a
plegamientos incorrectos de la proteína y en última instancia a su precipitación en los
denominados cuerpos de inclusión que se observan en el núcleo y citoplasma de las células
afectadas de los pacientes. En los cuerpos de inclusión no sólo se encuentra la proteína
Huntingtina mutada y normal, sino también otras proteínas que han sido “secuestradas”
por su precipitación, y que, por tanto, tampoco podrán ejercer su función normal en la
célula.
Los sistemas celulares responsables de la degradación de las proteínas mal plegadas dejan
de funcionar correctamente con la edad. Esto hace que al envejecer aumente el número de
cuerpos de inclusión en las neuronas, explicando la aparición tardía de la enfermedad.
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 12PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
DIAPOSITIVAS 22-25. Enfermedad de Huntington
4.2.-Herencia Autosómica Recesiva
La herencia autosómica recesiva afecta a alrededor de 2-3 individuos de cada 1000. La
principal característica del árbol genealógico es que la enfermedad no aparece en todas las
generaciones. Debido a su carácter autosómico, este tipo de herencia se manifiesta tanto
en hombres como en mujeres y ambos tienen el mismo riesgo o probabilidad de
transmisión a sus hijos e hijas. Por regla general, los individuos que manifiestan la
enfermedad son pocos y no suelen tener un progenitor afectado. Además, debido al
carácter recesivo, los individuos enfermos presentan las dos copias del gen mutadas
(homocigotos) y sus progenitores son heterocigotos (portadores).
Como los alelos mutantes son, en general poco frecuentes en la población, la mayoría de
los enfermos son heterocigotos compuestos, es decir tienen mutaciones diferentes en cada
alelo.
Los emparejamientos consanguíneos, en los que los miembros de la pareja tienen una
relación de parentesco, incrementan el riesgo de las enfermedades de herencia autosómica
recesiva. La probabilidad de encontrar en la población dos heterocigotos para este tipo de
enfermedades poco frecuentes es muy rara. Sin embargo, si ambos son de la misma familia,
la probabilidad es mayor, ya que comparten más genes. Así, cuanto más rara es una
enfermedad recesiva, más probable es que los padres de un afectado tengan algún tipo de
parentesco genético, algún ancestro común.
La predicción genética en familias con enfermedades que tienen herencia autosómica
recesiva depende del tipo de pareja estudiado (ver diapositiva 27). Algunos ejemplos son:
Dos progenitores sanos sin antecedentes familiares. La probabilidad de tener hijos
afectados depende de la frecuencia de los heterocigotos en la población. La
probabilidad será:
h (frecuencia portador en la población) x h x 1/4 = h2/4
Uno de los padres afectado y el otro sano, sin antecedentes. La probabilidad será:
h x 1/2 = h/2
Progenitores sanos pero uno con antecedentes, por ejemplo un hermano afectado.
La probabilidad será:
h x 2/3 x 1/4 = h/6
Pareja portadora de una enfermedad autosómica recesiva, que ha tenido un hijo
enfermo. La probabilidad de tener otro hijo enfermo es ¼, y cada hijo es
independiente.
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 13PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
Pareja que han tenido un hijo enfermo, ¿qué probabilidad tiene uno de los
progenitores de tener otro hijo enfermo con otra pareja que ya ha tenido 6 hijos
sanos?
Utilizamos la probabilidad condicionada, en la primera pareja los dos son portadores, pero
en la segunda pareja, el cónyuge tiene la probabilidad poblacional de ser portador. La
probabilidad condicionada modifica esta probabilidad a priori porque ya ha tenido 6 hijos
sanos.
Muchas enfermedades recesivas son metabolopatías, que afectan a enzimas de rutas
metabólicas. La mitad de la dosis de la enzima suele ser suficiente para realizar la función
biológica, así, los heterozigotos son portadores, pero no están afectados. Si hay un bloqueo
en una ruta enzimática, puede ser que haya una ausencia del producto, o que el sustrato se
acumule y se metabolice por vías alternativas dando lugar a productos tóxicos. La
manifestación clínica de una enfermedad puede ser debida a una cosa u otra.
Se observa homología fenotípica, es decir, se dan fenotipos compartidos por enfermedades
debidas a deficiencias enzimáticas que actúan en el mismo proceso metabólico.
Por ejemplo, en el albinismo se dan mutaciones en el gen de la tirosinasa, que oxida la
tirosina para formar melanina. De esta manera no habrá melanina (producto).
Los casos de la fenilcetonuria o de la galactosemia son ejemplos de acumulación del
sustrato. En la fenilcetonuria se acumula fenilalanina en sangre, que puede metabolizarse
en por rutas alternativas, dando lugar al ácido fenilpirúvico, muy tóxico para el desarrollo
del sistema nervioso.
Ejemplos de enfermedades autosómicas recesivas (diapositiva 28):
Albinismo
Fibrosis quística
Ataxia de Friedreich
Fenilcetonuria
DIAPOSITIVAS 25-29 Herencia autosómica recesiva
Fibrosis quística como ejemplo de enfermedad autosómica recesiva
Es la enfermedad hereditaria, autosómica recesiva, con mayor incidencia en la población
de origen europeo (1/2000-2500 recién nacidos). Con una frecuencia de portadores de
1/20-25 personas en Europa. Esta prevalencia ha hecho que se incluya dentro del cribado
neonatal. El código OMIM es # 219700, donde el 2 indica herencia autosómica recesiva.
La fibrosis quística se produce como consecuencia de mutaciones en el gen CFTR que
provocan una pérdida de función. CFTR codifica para la proteína reguladora de la
conductancia transmembrana de la fibrosis quística (CFTR, por sus siglas en inglés). CFTR
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 14PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
es un canal de cloro, que permite el paso del ion cloruro y bicarbonato a través de las
membranas celulares, y se localiza en la zona apical de muchos epitelios glandulares. Su
deficiencia altera la producción de sudor, jugos gástricos y moco.
La proteína está formada por dos dominios de membrana, cada uno de ellos con 6
dominios transmembrana (ver diapositiva 31) que se embeben en las membranas
celulares y forman el canal. Después hay dos dominios de unión a ATP necesarios para
abrir y cerrar el canal. Y finalmente encontramos un dominio regulador que es el que
determina cuándo el ATP puede unirse, y que se activa al ser fosforilado. En la actualidad,
existen más de 1.500 mutaciones descritas para este gen. Algunas mutaciones son más
frecuentes que otras, como la deleción de la fenilalanina en el codón 508, responsable de
hasta un 70% de los casos en la población europea. Se cree que esta frecuencia tan alta es
debida a que en algún momento ha proporcionado una ventaja selectiva frente al cólera y
al tifus. En ratones se ha visto que los heterocigotos son más resistentes al cólera, porque
las toxinas del cólera no inducen la misma pérdida de agua que en una célula normal, y por
tanto están protegidos contra la deshidratación. En el caso del tifus, la bacteria utiliza el
canal iónico afectado para translocarse dentro de la célula. Los pacientes heterocigotos
para la mutación translocan mucho menos la bacteria y los homocigotos no translocan
nada.
Encontramos 6 clases en función de como la mutación afecta a la proteína (ver diapositiva
32), que dan información de la gravedad de la enfermedad. Aunque al actuar también
genes modificadores, la relación entre mutación y efecto no es tan directa. Todas las
mutaciones descritas tienen como consecuencia una pérdida de función, a) impidiendo la
síntesis de la proteína, b) impidiendo el correcto plegamiento de la proteína, lo que
provoca que no se desplace a la membrana, c) alterando la unión del ATP, lo que causa que
el canal no sea funcional, d) afectando a los dominios de las paredes del canal, lo que hace
que el intercambio de aniones cloruro no sea tan eficiente, e) sintetizando menos proteína
de lo normal, lo que hace que haya menos canales, f) haciendo que la vida media de la
proteína sea muy corta. Las situaciones más graves son en las que no existe canal o cuando
este no es funcional.
La consecuencia de la alteración de la producción o función de la proteína CFTR es que el
transporte de líquidos y electrolitos a través de las membranas de los epitelios glandulares
se desequilibra. En los pulmones se forma una mucosidad espesa y con poca agua que
recubre la mucosa del epitelio pulmonar, impidiendo la aireación y constituyendo un nicho
para la infección de determinadas bacterias como pseudomonas, lo que causa neumonías
recurrentes. En el caso de las glándulas sudoríparas, el cloruro de sodio no puede pasar a
través de la membrana celular y se acumula en el sudor, dando lugar al característico sudor
salado de los pacientes. En el páncreas la mucosidad espesa obtura los conductos que van
al aparato digestivo, y no se pueden verter a este tejido las enzimas pancreáticas. Como
consecuencia las enzimas retenidas en el páncreas forman quistes y el enfermo presenta
desnutrición, porque no puede digerir bien los alimentos. También causa esterilidad en
varones, porque no se forman los conductos deferentes durante el desarrollo embrionario,
debido a que la mucosidad colapsa totalmente los conductos de Wolf. Las mujeres también
presentan fertilidad reducida por la gran cantidad de mucosidad acumulada en el interior
del útero.
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 15PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES GENÉTICAS
Material didáctico: Módulo 1 - Clase 2
DIAPOSITIVAS 30-32: Fibrosis Quística
4.3.-Herencia ligada a los cromosomas sexuales.
Los cromosomas sexuales son los identificados como X e Y. Las mujeres presentan dos
copias de X y los hombres tienen una copia de X más una copia de Y. Los cromosomas
sexuales no sólo contienen los genes que determinan el sexo, sino que también presentan
otros genes que influyen sobre ciertos caracteres hereditarios no relacionados con el sexo.
El cromosoma X contiene 1100 genes y el cromosoma Y solo 50.
Aunque los cromosomas sexuales constituyen un par de cromosomas homólogos (XX en la
mujer y XY en el hombre), no hay una identidad genética completa: en el par XY ambos
cromosomas presentan genes particulares y exclusivos en el denominado segmento
heterólogo (llamado también diferencial o no homólogo). La porción restante de los
cromosomas del par XY corresponde al sector homólogo. Así, los varones llevan un sólo
representante de cada gen de aquellos localizados en el sector heterólogo del X.
Los genes de la región específica de X van a presentar una herencia ligada al cromosoma
X (también llamada herencia ligada al sexo), y los genes situados en esta región específica
de Y van a presentar una herencia ligada al cromosoma Y (también llamada herencia
holándrica). Por último, los genes presentes en la región homóloga entre X e Y, presentan
u n a herencia parcialmente ligada al sexo o pseudoautosómica. Estas regiones de
homología se focalizan en los extremos de ambos cromosomas (regiones PAR1, con 2.6 Mb
y 24 genes y PAR2, con 320 Kb y 4 genes), pero también se pueden encontrar pequeñas
regiones homólogas dispersas por los cromosomas. Los cromosomas que dieron lugar a X
e Y eran como un par de autosomas, con la evolución uno de ellos ha reducido su tamaño y
ha ido acumulando secuencias de ADN repetitivo. Los genes que comparten hoy en día son
una reminiscencia de ese antepasado común.
Los hombres transmiten el cromosoma X a todas sus hijas y el cromosoma Y a todos sus
hijos, mientras que las mujeres pueden dar cualquiera de sus cromosomas X a sus hijas o a
sus hijos.
Dado que las mujeres presentan dos copias de X, podrán ser homocigotas o heterocigotas
para un gen con varios alelos en X, mientras que los hombres, al tener una única copia de
X, son considerados hemicigotos y expresarán la condición indiferentemente de que esta
sea recesiva o dominante. Por esta razón, para las enfermedades recesivas ligadas a X,
habrá muchos más hombres afectados que mujeres, y para las enfermedades dominantes
ligadas a X, encontraremos tanto hombres como mujeres afectados, pero los hombres
estarán más gravemente afectados que las mujeres.
La mayoría de los genes se encuentran en la región específica de X, por eso a la herencia
ligada a X se le llama comúnmente Herencia ligada al Sexo.
Título de Experto Universitario en Medicina Genética y Genómica 2019 Página 16También puede leer

















































